Yesterday’s post updated my diagram showing relationships between PSP/CBD and the other major neurodegenerative diseases. The only substantive change was the addition of a tauopathy called “anti-IgLON5 syndrome,” which we’ll call “AIS.” It’s not “major” in the sense of “common,” but in the sense of “important to know about because it’s treatable, especially if diagnosed early.”
AIS sits on the border between the autoimmune and the neurodegenerative diseases. (In fact, multiple sclerosis does that, too, but unlike most neurodegenerative diseases, it has no known protein aggregates in the affected brain cells.) In AIS, the blood and/or spinal fluid have antibodies directed against one type of the “cell adhesion molecules” on the surface of brain cells. Those molecules assist in the function of the microtubules, which are the cell’s internal skeleton and transport system, and also where tau normally works. The problem is that we don’t yet know for sure whether the antibody attack causes the cell damage or if the antibodies are merely the immune system’s reaction to damage caused by something else.
The classic symptoms of AIS are major difficulties in the control of sleep and involuntary movements called “chorea.” But more recently, four different types of AIT have been described, and one of them mimics PSP, with severe loss of balance and milder cognitive loss and predominantly vertical eye movement problems. As in ordinary PSP, sleep problems are present as well, but with more dream enactment and obstructed breathing than occur in PSP.
About a quarter of those with AIT have the PSP type. The other three types emphasize problems with sleep (the most common); speech and swallowing; and cognitive problems, respectively. All four types also display autonomic disturbances in a majority of patients, including episodes of sweating, incontinence, and slow or fast heartrate. Oddly, only a few have low blood pressure. Otherwise, the autonomic features are similar to those of MSA, and of course the motor features of MSA, like those of PSP, can be similar to those of AIS, but without the chorea.
An important difference between AIT and PSP, CBD or MSA is that AIS has no muscle rigidity, movement slowness or tremor. Those three things are collectively called “parkinsonism.”
AIS’s average onset age of 64 plants it firmly in the range of most neurodegenerative diseases, rather than in the younger range typical of autoimmune diseases. Furthermore, the female predominance of most autoimmune diseases does not exist for AIS. Also, only one case in the literature has been reported to spontaneously improve, and the course is slowly progressive rather than fluctuating. These points favor a neurodegenerative origin.
On the other hand, all patients with AIS have a specific genetic variant in one member of a set of genes on chromosome 6 associated with autoimmune disease. It’s called the human leukocyte antigen, or HLA system. And here’s the most important point favoring an autoimmune origin: treatment with standard immune modulating drugs such as steroids, intravenous immunoglobulin and azathioprine, helps about two-thirds of the patients.
It’s interesting that the classic form of AIT, where sleep disturbances predominate, is much less responsive to immune modulatory drugs than the more recently-described variants. So maybe the different types sit slightly on one side or the other of the autoimmune/neurodegenerative fence.
Here’s a colorful diagram showing features in 22 patients with AIS. It’s from the research team in Barcelona that and first described AIS in in 2014 and is a world leader in neuro-autoimmune disorders.
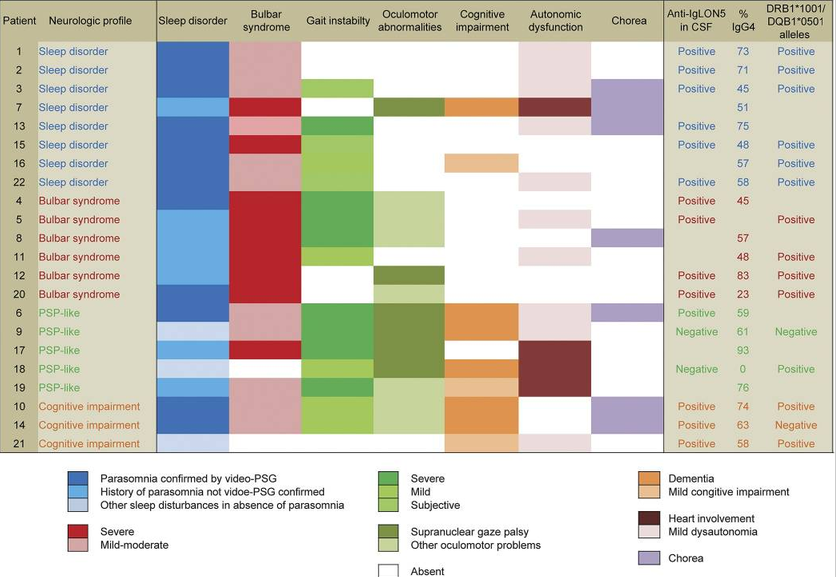
This diagram doesn’t mention that some patients with AIS exhibit hyperexcitability in the form of muscle cramps, muscle jerks and easy startle.
I must emphasize that while AIS is a tauopathy, it’s not PSP or CBD. It affects different parts of the brain, has both 3R and 4R tau (PSP and CBD have only 4R), it has a longer survival untreated, and it has anti-IgLON5 antibodies.
So here’s the take-home:
- People in the early stages of an illness suspected of being PSP, CBD or MSA should make sure their doctor knows about AIS so that testing for the antibody can be considered. If it’s positive, immunomodulating treatment may make a big difference, especially if two or more such drugs are used together, and if treatment is started in the first two years of the illness. Often, rheumatologists know more about the treatment of autoimmune disorders than neurologists, especially movement disorder specialists.
- Even if there’s no response to immunomodulatory treatment, finding anti-IgLON5 antibodies should prompt a search for small but growing cancer. While none of the publications on AIS has examined this issue thoroughly, other autoimmune disorders in the nervous system are very often associated with cancer. Most of the published case series either didn’t work up the patients for cancer or lost follow-up before a small cancer would have revealed itself. Detection and removal of a small, early cancer could help the AIS, and even more important, save a life.


{kind=link}