As promised, I will now start trying to keep you updated on actively recruiting clinical trials in PSP. Keep in mind a few things before using my list:
In compiling my list, I rely heavily on www.clinicaltrials.gov. Trials in the US are required to list themselves there, but some take longer than others to comply. Trials overseas may or not be required to have a listing on clinicaltrials.gov, depending on the country. My list also includes trials I’ve heard about through the grapevine that are not (yet) listed.
In Phase 1 and sometimes in Phase 2, the trials do not typically allow patients completing the protocol to continue receiving the drug, no matter how well they were doing on it. Later Phase 2 and Phase 3 trials often do allow this, but they do not commit to it in advance. Typically, if the trial shows the drug to be ineffective (or unsafe) overall, the drug company will discontinue its program for that drug and stop producing it. That means that even the few patients who might have been doing well on it will not continue to have access to it.
In theory, exceptions to that rule could exist for drugs that are being tested in both PSP and another disease such as Alzheimer’s. If the PSP trial shows lack of efficacy but acceptable safety, the company will continue to manufacture the drug pending the outcome of the other trial and may make it available to those who completed the PSP trial.
For trials of neuroprotective treatment, patients may not know if they are benefitting. (Definitions: “Neuroprotective” drugs are intended to slow the progression of the abnormal brain process in the long term, as opposed to helping the symptoms experienced by the patient in the short term. For example, for infections, antibiotics are protective, while painkillers are symptomatic.) To know if an individual in a neuroprotective trial has had slowing of their rate of worsening over the 12 months on the drug, their rate of worsening over at least a few months before the trial would have to have been quantitatively assessed using the same method that the trial used. We almost never have that data, so trials work by comparing the average result from a group receiving the drug to the average from a group receiving a placebo.
Do not expect a neuroprotective drug to improve your symptoms. At best, it could prevent worsening, and more likely would only slow the rate of worsening. The trials are typically designed to detect a slowing of the rate of worsening of 30% or 40%.
Bottom line: Participating in a clinical trial, especially one in Phase 1 or 2, requires some level of altruism.
For more information on these trials, go to www.clinicaltrials.gov and enter the drug’s name or ID number shown in the first column.
Here’s the current list to the best of my knowledge. The ones at the bottom labeled as “may start” are awaiting funding in most cases.
I’ve been writing about PSP for patients and families for 30 years, and I realized long ago that what people most want to hear about isn’t my scientific musings about a new diagnostic technique or etiologic theory. They want to know about treatment, especially about clinical trials they might join.
For most of those three decades, there was little to report on that front. But in the past five to eight years, that has changed. Fortunately, clinicaltrials.gov has stepped into the breach starting with the FDA’s 2007 requirement that pharmaceutical companies list their trials on that public bulletin board.
But clinicaltrials.gov has its drawbacks for patients and families seeking a treatment trial. Searching its database on “progressive supranuclear palsy” returns a long list of projects that are mostly either observational, geographically unrealistic, fully recruited, terminated, listed but nowhere near initiation of recruitment, or on hold because of the pandemic. Yes, careful scrutiny can eliminate those, but that takes some insight into clinical research that most people lack.
I haven’t been a complete slacker on this matter. I wrote a 2018 book entitled, A Clinician’s Guide to Progressive Supranuclear Palsy, but that was frozen in time. I led the July 2021 writing, with 36 expert co-authors, of a consensus statement entitled, Best practices in the clinical management of progressive supranuclear palsy and corticobasal syndrome: a consensus statement of the CurePSP Centers of Care. It focused on available, non-specific, symptomatic management, with only vague predictions about what actual disease-specific, neuro-protective or preventive treatments might be on the horizon.
That’s why this blog will henceforth try to keep you all current on clinical trials in PSP and CBD. I’ll do this in concert with CurePSP, which will soon add such a page to its website, which the CurePSP staff and I will update as needed. I’ll get back to you on this soon.
Since 2014, I’ve been trying to find the cause of a geographical cluster of PSP in northern France. It’s the only documented PSP cluster known. The problem was difficult enough, but now the cluster has mysteriously disappeared.
The clinical aspects of the cluster are detailed in this 2015 paper. Here’s the executive summary: In 2005, Dominique Caparros-Lefebvre, MD, a geriatric neurologist with experience in PSP research, arrived at her new practice position in Wattrelos, France, an industrial suburb of Lille. By 2007, she started to notice more PSP than expected and developed an excellent database. She diagnosed 100 patients over the next decade. In 2013, she invited me to help her find the cause, as I had had some experience in the epidemiology of PSP. I calculated the observed-to-expected incidence ratio of PSP to be 12.3 in Wattrelos and its neighbor to the south, Leers. Most clusters of chronic diseases such as cancer have ratios much lower, in the range of 1.5 or 2.0. So this was a major cluster.
PSP in the 100 patients has differed only slightly from “sporadic PSP,” with more PSP-Parkinsonism than PSP-Richardson syndrome, and an older mean onset age, 74.3. The 13 autopsied cases show typical PSP, with the expected 4R tau and the H1/H1 genetic haplotype. That work was done by a very accomplished research team at the University of Lille led by Luc Bueé, PhD and Vincent Deramecourt, MD, PhD. No other molecular genetic workup has been performed to date, but none of the affected persons were related to one another and among the patients are 7 Algerian immigrants, a strong point against a genetic origin.
Wattrelos and Leers have extensive chemical contamination, especially by metals from an ore extraction plant that operated in southern Wattrelos for most of the 20th century. Huge piles of spent chromate and phosphate ore, now covered, remain on the plant’s property, which has been converted into to a public park after mitigation efforts between 2000 and 2010. Only 2 of the 100 patients worked in the chromate/phosphate ore plant, but soil from the area adjacent to the slag heaps has been used as fill in construction and road maintenance over a wide area. Furthermore, multiple chemical-related industries such as tanning and dyeing formed the base of the town’s economy for many decades.
So, the obvious culprit has been metals. Chromium is a carcinogen but not a good candidate as a direct neurotoxin, as its most common form, hexavalent chromium, does not cross the blood brain barrier. Nor is phosphorus a good candidate, but phosphate ore often contains important levels of other metals.
In France, growing one’s own herbs and vegetables is a common practice, even in densely urban areas. Dr. Caparros suspected thyme, a widely used herb in French cooking that avidly absorbs metals from the soil. The French government’s soil data and our analysis (by my Rutgers colleague Brian Buckley, PhD) of home-grown thyme samples from Wattrelos suggested that arsenic, cadmium and nickel were the most likely possibilities.
In 2016, I recruited a team of neuroscientists led by Aimee Kao, PhD of UCSF, with skills in stem cell models of PSP and access to stem cells with PSP-related tau gene mutations. As an initial project, they created brain cells with a rare PSP risk mutation (to create a “background” vulnerability) and exposed them to chromium, cadmium and nickel. They did the same experiment with cells from the same PSP patient except that the PSP risk mutation was converted to normal using CRISPR. They found that some of the same damage seen in PSP — aggregation of tau and evidence of apoptotic (i.e., programmed cell death) in the exposed cells with the mutation. But those abnormalities are not specific for PSP. We published that in 2020 and unfortunately, I couldn’t keep that team together for follow-on projects. Equally unfortunately, the local French human research authority would not allow Dr. Caparros to perform further field work that might have pointed to a specific metal and route of exposure.
So why aren’t clusters of PSP seen in the many other places in the world where those metals contaminate the environment? My own pet theory was that toxicity from multiple metals acting in concert is needed, and no place other than Wattrelos/Leers has a combination of so many metals in one spot together with a physician able to diagnose PSP as well as Dr. Caparros. So, one of the follow-on projects might be to repeat the lab experiments with combinations of the same and other metals that are known to occur in the environment, either in Wattrelos/Leers or elsewhere, either as a result of industrial pollution or naturally occurring.
As I was starting to make plans for such a project with lab colleagues at Rutgers, it became clear that the number of patients whose onset occurred since 2013 has been declining. The most recent onset date of the 100 patients is 2016. This is not how a cluster of toxic (or genetic) cause should act. It’s possible that the mitigation efforts on the two slag heaps reduced the rate of entry of chromium and phosphate into the local environment, but the metals were pervasive in the area and presumably remain so.
—-
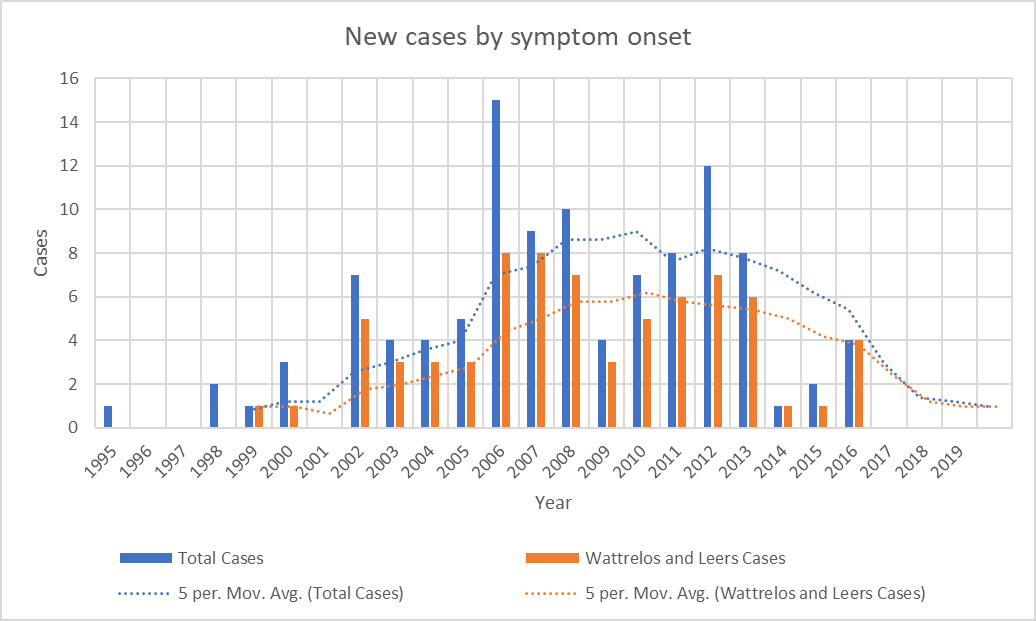
Graph showing the disappearance of cases with onset since 2016. Bars show the number of cases with onset in each year. Dotted lines show the average over the previous 5 years. “Total Cases” refers to all cases in Wattrelos and Leers as well as in nearby towns where patients were likely to use the Wattrelos hospital. The paucity of cases with onset before 2002 is explained by the arrival of Dr. Caparros in Wattrelos in 2005. Before that, no physician likely to have been able to diagnose PSP worked there and few patients with onset before 2002 would have survived to come to Dr. Caparros’ attention. Still, we cannot rule out the possibility that the cluster in fact started in the 1990s and its disappearance by 2017 is consistent with this possibility.
—-
The unexplained abatement of a geographical or temporal disease cluster speaks for an infectious cause. The salient example of an infection causing a temporal cluster of a neurodegenerative tauopathy is postencephalitic parkinsonism (PEP). That cluster started 2 years before and ended a decade after the great “Spanish” flu pandemic of 1918-1920 and is independent from it. PEP was a chronic, levodopa-responsive parkinsonism that affected people of any age who recovered from an encephalitis that was presumably viral, but the specific virus has not been identified. At autopsy, the brain showed neurofibrillary tangles not very different from those of PSP. The last patients with PEP died before modern molecular techniques were available, so its cause may never be known.
Could the cause of the Wattrelos/Leers cluster have been a virus? True, there seemed to have been no antecedent encephalitis, but it may have been mild, self-diagnosed as a cold or the flu, and forgotten by the time Dr. Caparros saw the patient decades later. But there need not have been any clear acute-phase symptoms at all. The virus could have set up a slow process of damage involving tau aggregation, starting with inserting its own genetic material into that of the host. Or the initial infection could have altered the patient’s immune system in a way that encouraged (or allowed) the pathology of PSP to develop. Let’s not forget that disordered immune modulation is one of the up-and-coming theories of PSP-causative factors.
If a virus contributed importantly to the cluster, could ordinary, “sporadic” PSP outside of the cluster be the result of a similar virus? Or maybe sporadic PSP is caused by the same virus without the predisposing local factor of the unusual metals exposure. Or maybe a virus infected the gut microbiome of the Wattrelos population in a way that increased PSP risk. I could go on.
We know that at least one neurodegenerative condition, sporadic Creutzfeldt-Jakob disease, is caused by an infectious agent (in this case the prion protein) without geographical or temporal clustering. The idea of a virus or prion as a cause of PSP is not new, and previous attempts to prove that hypothesis starting in the 1970s have been negative. But the technology for finding viral fingerprints has improved markedly since then.
I’ll try to get some of the research honchos I know interested in this theory and get back to you.
Here’s something that may seem too good to be true, and in my opinion, it is. I’m writing about it as a cautionary measure.
Researchers at Guangzhou University, China have published a case report of a man with advanced PSP who received intravenous and intrathecal (into the spinal fluid) infusions of stem cells derived from umbilical cord blood. His symptoms had started unusually young, at age 53, and 8 years later, his PSP Rating Scale score was 73 – a typical score for that duration of disease. He received the infusions at that point and has survived another 8 years to date with essentially the same PSPRS score.
Although the patient is (apparently, but not explicitly) still alive and there has been no autopsy to prove the accuracy of the diagnosis, the clinical history, neuro exam and published MRI images are typical for PSP. The subtype is probably PSP-Parkinson, as his falls didn’t start until 3 years in and he didn’t need gait aids until 2 years after that. The authors state that the subtype was PSP-Richardson syndrome because the patient met those criteria at the time they first saw him, 8 years in. But PSP-P usually evolves into PSP-RS in the advanced stages. The life expectancy of PSP-P with typical onset age in the 60s is about 10 years, and for those with onset as young as age 53, would be a few years more. So survival (to this point) of 16 years is not far from expected.
But that’s not the main problem. The main problem is that the PSP Rating Scale has a “ceiling effect.” That is, patients progress at an average rate of 10 or 11 points per year, and once the score reaches the 70s, it stops progressing although the patients continue to survive and to worsen. That’s because the worst possible score on many of the scale’s 28 items doesn’t capture the full level of dysfunction that can yet occur. Another reason for the ceiling effect is that some of the 28 items don’t affect some patients severely or at all, even late in the disease course. Examples are dystonia, tremor, irritability, emotional incontinence, horizontal eye movement loss, and limb apraxia. So the patient of Dr. Li et al may simply have reached his PSPRS ceiling and continued to survive by virtue of the unusually good general care that study subjects typically receive.
There are some other issues:
There’s no objective evidence that the infused cells survived.
There’s no measure of any sort of growth factor in the blood or CSF that might provide a mechanism of action of stem cells in halting an otherwise rapidly progressive disease in its tracks.
There’s no functional measure of brain metabolism such as fluorodeoxyglucose PET or magnetic resonance spectroscopy to objectively document a halting/slowing of progression.
The 2 MRI were performed at the time of the infusions and 2 years later. Although the authors claim “no deterioration” in the MRI over that time, the 2 sets of images do show progression of atrophy, both to my eye and by the formal measurements superimposed on the images. The midbrain’s diameter on the before-and-after sagittal images (Fig. 1, images A-1 and B-1) declined from 11.84 mm to 10.64 mm and the pons from 21.07 mm to 19.37 mm. Both are typical for PSP. The before-and-after axial images (images A-2, A-3, B-2 and B-3), where the measurements seem to indicate some improvement in the atrophy, are performed different scanning planes. That can seriously affect the simple measurements performed. In fact, comparing the 2 sagittal images shows that the patient’s head is at 2 very different tilt angles in the scanner. That problem affects the measurements in the axial plane but not in the sagittal plane.
As Dr. Li and colleagues point out, “randomized controlled trials are needed in the future . . . “ I would advise people with PSP not to undergo this, or any, experimental procedure outside of a formal trial at a reputable academic institution. If you’re considering it, make sure the study is listed in clinicaltrials.gov, although that by itself no guarantee of anything. Make sure that the researchers have a track record of peer-reviewed publication in this area. If the doctors doing the treatment are willing to use it for a wide variety of unrelated disorders, be suspicious. If the claims omit mention of side effects or toxicity, be doubly suspicious. If there’s no mention of success in any sort of animal model, that could be a problem. Finally, if a “research study” charges a hefty fee, stay away. (We have no evidence that this was the case for Li et al.)
We should encourage fresh ideas for the treatment of PSP and other neurodegenerative diseases. We should not be biased against research from countries with still-developing research infrastructure and institutional safeguards. But we should also know how to evaluate the quality of research reports and to be vigilant for signs of quackery.
If you like my research updates but need a break from the molecular stuff, here’s an interesting and unexpected clinical discovery, usable at routine visits for care of PSP.
Michelle Troche, PhD, is a speech/language specialist in the Laboratory for the Study of Upper Airway Dysfunction in the Department of Biobehavioral Sciences at Columbia University’s Teachers College. She’s a past grantee of CurePSP’s
The two groups of 26 patients were designed to be similar in terms of age, sex, disease duration and severity of swallowing difficulty. Their cough was analyzed with a device called a pneumotachograph (“air-speed-writer’) that fed the data into a software system. The procedure was performed during both coughing on request and coughing induced by a 2-second spray of capsaicin in 4 progressively increasing concentrations. Capsaicin is an irritant found in hot peppers, so kudos to those volunteer patients!
After a sophisticated statistical correction for confounding factors, some results were as expected: that the patients with PSP were able to generate only a fraction of the expiratory flow rate and volume of those with PD. However, another result was unexpected: patients with PSP were more bothered by increasing concentrations of the capsaicin than were those with PD, but were no more likely to respond by coughing.
In the researchers’ words, “. . . it is interesting to note that although both groups exhibited blunted urge-to-cough slopes compared to prior research in healthy adults, patients with PSP demonstrated increased urge to cough compared to PD. This means that even though the participants with PSP were perceiving the increasing cough stimulus more than patients with PD, they were not coughing more to that stimulus.“
So what does this mean? We think of PSP as a motor and cognitive disorder, but this result shows that there’s also a sensory deficit. The sensory input into the cough reflex takes place in the brainstem, the main location of pathology in PSP, so this result makes sense. It’s just that no one had previously thought about it. (The same phenomenon accompanies many important scientific discoveries – they seem so obvious in retrospect.)
Like any pioneering work, this one has its limitations. Although the PSP and PD groups were similar in terms of disease duration (both about 5 years) and swallowing function, the PSP group was much worse in terms of overall disability as measured by the widely-accepted Schwab and England Activities of Daily Living (SEADL) scale. With 100 being normal on the SEADL, the PSP group averaged 48, the PD group 79. This is expected given PSP’s more rapid disease course from onset to death — the 5-year disease duration is a far greater fraction of the average survival in PSP (7 years) than in PD (15 years). So perhaps the authors should/can add the SEADL score to their statistical model. Another issue is that the experimental procedure did not consider any effect on the cough function of PD medication, which dramatically helps general motor function of PD but not of PSP.
Now, how can we use this information? Dr. Troche and colleagues suggest that patients with PSP be monitored closely for cough deficit and that their own, previously published protocol for sensorimotor training in PSP, referred to above, could be instituted sooner rather than later.
Most of my posts are long — maybe too long. The charitable explanation is that I can’t resist my instincts as a professor to explain stuff so my learners can understand it. The less charitable explanation is that I’m just a windbag. So here are a bunch of very brief items of news, ideas and opinion about PSP and CBD in the style of Twitter. In fact, I’ll even limit my character count to 280, including spaces. Here goes.
A group in Bologna did skin biopsies to look for a phosphorylated form of α-synuclein in PD, PSP or CBD, and controls – 26 subjects in each group. They found it in all 26 with PD, in no controls, and in 24 with PSP/CBD. (The other two had PD-like features.) Now: how about MSA?
You’ve noticed that CurePSP’s publicity materials call PSP, CBD and MSA “prime of life” diseases because those conditions’ usual decades, the 50s, 60s and 70s, are when life can otherwise be lived to the fullest. Do you agree? Let me know.
A group called the PSP Research Roundtable was formed in 2017 to help speed the process of testing promising drugs. It’s run by CurePSP and has membership from academia, the FDA, the NIH, drug companies, biotech, philanthropy and patient advocacy groups.
Transposon Therapeutics has started a Phase 2 trial of TPN-101 in PSP at private clinical trial sites in Boca Raton, FL and Farmington Hills, MI. Like many available HIV drugs, TPN-101 inhibits the enzyme reverse transcriptase, but otherwise, details are sparse.
About the mechanism of action of TPN-101: I can tell you that another reverse transcriptase inhibitor routinely used for HIV called efavirenz (trade names Sustiva and Stocrin) reduces tau aggregation. CurePSP is currently supporting a study of it in a mouse tauopathy model in The Netherlands.
You’ve heard me whining that we need a diagnostic “trait marker” for PSP. In other words, we need to be able to accurately distinguish PSP – during life — from such mimics as Parkinson’s, multiple system atrophy, Alzheimer’s, corticobasal degeneration, normal-pressure hydrocephalus and others. Only in that way can we create “pure” groups of patients in which to study the disease and test specific treatments.
Right now, the best diagnostic test we have is the MDS-PSP Diagnostic Criteria, which requires only traditional history-taking and a hands-on neurological exam. Those criteria work well for PSP-Richardson syndrome after the first couple of years but not quite well enough for earlier-stage PSP-RS nor for the “minority” or “atypical” types, which together account for 60 to 75 percent of PSP.
The most promising markers using laboratory or imaging data are levels of phosphorylated tau and neurofilament light chain (NfL) in the spinal fluid and blood; and perhaps MRI measurements of the size of the midbrain, pons and related areas at the base of the brain. But these are far from ready for prime time. NfL is a protein component of brain cells that has been shown to occur at about a two-fold higher level, and to increase faster over time, in PSP and MSA than in PD, Alzheimer’s and other neurodegenerative diseases. MRI changes don’t occur in the early stages. Positron emission tomography is coming along, but won’t be ready for use in PSP for another few years, and even then will be costly and not widely available.
Last week, my routine surveillance of new PSP-related research papers in the literature yielded two interesting hits — both about PSP trait markers, both using new lab techniques, and both from Italy.
Corinne Quadalti and colleagues at the University of Bologna measured NfL and alpha-synuclein in spinal fluid and blood. They found that plasma NfL alone worked very well in distinguishing PD from PSP, with an accuracy of 0.94. (“Accuracy” in this context is the area under the receiver operating characteristic curve, which compares sensitivity with specificity. Perfect accuracy is 1.0 and a useless test’s accuracy is 0.5, where a coin flip would work as well.)
Alpha-synuclein is the main protein aggregating in Parkinson’s, dementia with Lewy bodies and multiple system atrophy. It is to those diseases what tau is to PSP and CBD. To measure it, they used a new technique called “real-time quaking-induced conversion” (RT-QuIC; pronounced, “R-T quick”), which measures that protein in its misfolded and aggregated forms. This prevents that abundant protein in its normal form from swamping the measurement. The result was positive in 91% of their patients with PD and in none of their 58 patients with PSP or CBD.
Now, if you have a nose for statistics, you’ll raise your hand and say, “But those 9% of PD patients with a negative test comprise more people in the general population than all the patients with PSP or CBD, so a negative test doesn’t mean much.” and you’d be right. So, while the sensitivity of the test for PD is excellent, the specificity is low, rendering the overall accuracy in a real-world situation insufficient.
For that reason, the authors combined two measurements – spinal fluid NfL and serum alpha-synuclein, with a resulting improvement in distinguishing PD from PSP/CBD to a sensitivity of 97.4% and specificity of 100%. That’s more like it, but keep in mind a few issues: They combined PSP and CBD into one group, and we don’t know if the results apply as well to each disease alone. They had no autopsy confirmation of the diagnoses, which means that these patients were already at a stage that was possible to diagnose using traditional clinical criteria; this means that patients with earlier-stage illness will be needed in a follow-up study. Finally, and as always, the results have to be confirmed at other centers using other techniques.
Exosomes are tiny bubbles of brain cell membrane enclosing whatever cell contents were there when the bubble pinched itself off and floated free. They often find their way into the bloodstream. MicroRNAs are stretches of RNA averaging only about 22 nucleotides. They do not encode proteins as messenger RNA (mRNA) does, but instead bind to mRNAs to regulate their translation into protein. They are specifically encoded in the DNA of the genome and about 2,000 of them are known to exist.
Dr. Manna et al measured levels of 188 miRNAs for which there is evidence of association with some neurodegenerative disease. They found a set of 6 miRNAs that together yielded an accuracy in distinguishing PSP from PD of 0.91. The accuracy for distinguishing PSP from controls was 0.90. Of course, many of the same caveats that I listed for the other paper apply to this one. Plus, PSP mimics other than PD were not included in the analysis. Just as important is that there were only 25 patients with PSP and they were a mixed bag of 20 with PSP-Richardson and 5 with PSP-Parkinson. In applying a marker for the purpose of excluding patients with PD from a study of PSP, it is critical to be able to distinguish PD from PSP-P. It is unlikely that those 5 patients with PSP-P constituted a statistically valid sample for that purpose. That will be a project for another day.
What do I take away from these two papers? Neither of them alone provides a marker just yet, and each has its drawbacks given the current early stage of work. But perhaps, with some refinement, combining them with other non-invasive markers could create a diagnostic panel with enough accuracy to distinguish PSP from all of its mimics. After all, in medicine in general, multiple diagnostic tests (several tests of body fluids, some imaging, a physiologic test such as an EKG) must be combined to produce an actionable diagnosis. Why should PSP be any different?
I think the problem (and it’s a good problem to have) is that new candidate markers are being identified all the time, as are ever more sophisticated technology for measuring them, with RT-QuIC, miRNA and exosomes as prime examples. That means as researchers turn their attention to early-phase development of newer ideas using newer technology, ideas that looked potentially useful if pursued further may be neglected and not developed into practical tests. What to do? Do we just let scientific nature take its course in its traditional, anarchical way, waiting for research groups to take techniques with good initial data to the next level? Or should a group of experts with an iron fist issue some sort of “white paper” listing which markers with good preliminary evidence, perhaps like the ones I describe here, should be nurtured with funding and collaborations? If so, who chooses those experts? And once the experts are chosen, how can we prevent them from favoring the ideas in which they’ve invested their own time, resources and reputations?