If PSP is an orphan disease, corticobasal degeneration (CBD) can’t even get into the orphanage. Like PSP, it’s a “pure 4R tauopathy”; it can resemble PSP in many cases; it leads to disability and death after a similar span of time; and it’s no more treatable. But its prevalence is about 10-20% that of PSP and it’s very difficult to diagnose in a living person. People fulfilling the accepted, published diagnostic criteria for the most common type of PSP (PSP-Richardson syndrome) actually have that disease at autopsy in over 90% of cases, but for CBD, the figure is less than 50%. That makes it hard to recruit a group of subjects for a drug trial — or any research — without other diseases influencing the result. That has put quite a damper on CBD research.
To add injury to injury, googling “CBD” reveals a lot more about cannabidiol than about corticobasal degeneration.
So, an objective diagnostic test for CBD would be great. Now, researchers mostly at Washington University in St. Louis (WUStL) and University of California, San Francisco (UCSF) have shown that two tiny fragments of the tau protein are less abundant in the spinal fluid of people CBD than in healthy people or those with PSP or three other rare tau disorders called argyrophilic grain disease, Pick’s disease and frontotemporal lobar degeneration associated with aggregation of TDP-43. They found no difference between CBD and Alzheimer’s disease or frontotemporal lobar degeneration with mutations in the tau (MAPT) gene, but in practice, those two disorders can be readily distinguished from CBD by other means.
The paper appears in the prestigious journal Nature Medicine and it’s open access, so I can provide you this file to download. The first author is Kanta Horie, PhD and the senior authors are Chihiro Sato, PhD and Randall Bateman, PhD, all of WUStL.
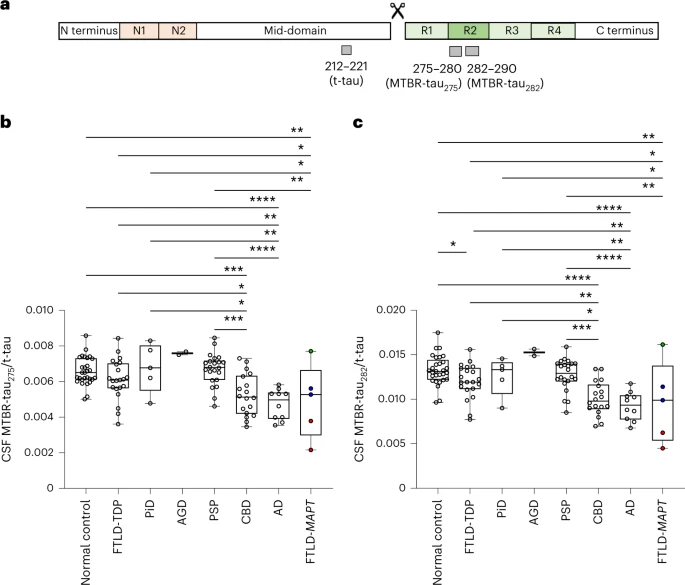
Panel “a” shows the tau protein. The four microtubule-binding domains are R1 to R4. The one whose inclusion or exclusion makes the difference between the 4R and 3R tauopathies is R2, which is encoded by the gene’s exon 10. The amino acids are numbered starting at the N terminus on the left. Two short stretches of amino acids, numbers 275 to 280 and 282 to 290, were the object of this paper’s analysis. N1 and N2 are two other sections, encoded by exons 2 and 3, respectively, that can be included or excluded in the finished tau protein.
Panel “b” shows the analysis of the 275-280 fragment of tau in the spinal fluid (CSF). The vertical axis is the ratio of the concentration of the 275-280 fragment divided by the concentration of total tau. The horizontal axis lists the tauopathies analyzed in this project. Each circle is one patient. The “box-and-whisker” plot shows, from top to bottom, the maximum value, the 75th percentile, the median, the 25th percentile, and the minimum value. The asterisks indicate the statistical significance of the comparison between the two groups at the ends of each horizontal line segment. One asterisk is a weak difference and four is the strongest. Pairs of groups without a horizontal line connecting them did not differ (i.e. the p value was greater than 0.05, meaning that any difference between them could have occurred by chance with a likelihood of more than 1 in 20).
Panel “c” shows the same thing, but for the 282-290 fragment of tau. The results are essentially the same as for the 275-280 fragment.
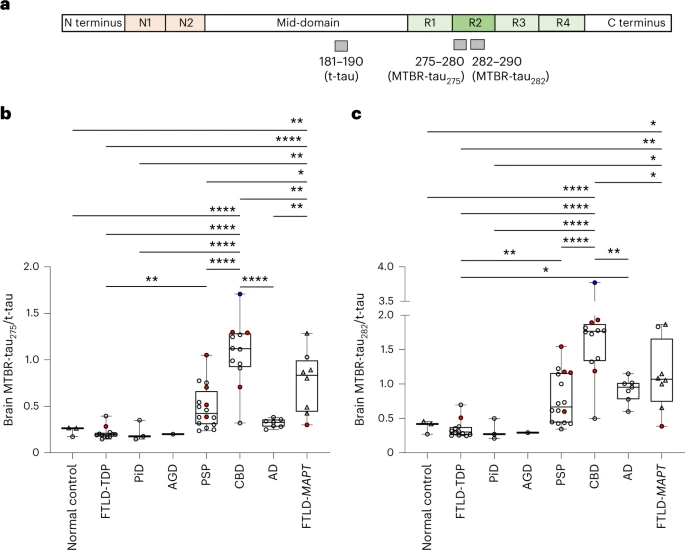
The odd thing is that the same analysis using autopsy brain tissue rather than spinal fluid gave a very different result: The values (i.e., the ratio of the fragment to total tau) was actually higher for CBD than for the other groups. The authors present various theories to explain this, but in any case, it does not detract from the diagnostic value of the spinal fluid results. Take a look and the brain tissue results:
So, what does this mean for people diagnosed with CBD, present and future? It means that if someone like a drug company has an experimental treatment that might help CBD, they could recruit a group of patients with a high level of confidence that they have excluded other diseases that could confound their results. That level of confidence is expressed as the “area under the receiver operating curve” or AUC. A previous post on this blog explains that statistic, which varies from 0.5 for a diagnostic test no better than a throw of the dice to 1.0 for a test that’s perfectly accurate every time. The AUC for this test to distinguish CBD from those other disorders (other than AD and FTLD-MAPT) is 0.800 to 0.889. That’s close to the figure for PSP using the neurological history and exam.
If this diagnostic test is confirmed (a big “if”) and enters use by researchers and drug companies, and if a drug company sees a route to profitability in so rare a disease, the only problem is finding enough patients with CBD for a trial. If CBD is 20% as common as PSP, and the new test for CBD is just as good as the present clinical diagnosis of PSP, then it will require five times the number of participating clinical test sites to fill a trial. But with international collaboration, it’s do-able.
Now, let’s hope that this test is adopted and that CBD is adopted.
