A disturbing piece of news this week about an influential 2006 paper in Nature about Alzheimer’s disease. Turns out it was likely that some of the data in the published version were deliberately faked. The paper was about beta amyloid, which is not an issue in PSP. In fact, this could actually be good news for PSP research. Here’s why:
In the experiments reported in the 2006 paper, researchers at the University of Minnesota Twin Cities used mice carrying a copy of the human amyloid precursor protein (APP) gene with a mutational variant known to cause AD in humans. (In the normal human brain, the protein product of the APP gene is cut to form beta-amyloid, abbreviated, “A-beta.”) The researchers allowed the mice to develop cognitive deficits, analyzed their brains, and found a type of small A-beta aggregates never before seen, dubbing them “A-beta*56.” They extracted the small aggregates, called “oligomers,” and injected them into the brains of genetically normal (“wild-type”) rats, which proceeded to develop AD-like cognitive disabilities.
Ever since A-beta was identified as a critical player in AD in 1984, researchers had been trying to nail down just what form that protein takes in the process of causing, or contributing to, the disease. The 2006 paper seemed finally to answer that question and formed the basis for innumerable subsequent experiments world-wide and hundreds of millions of dollars spent by the NIH, philanthropies and drug companies to build upon it in pursuit of an AD treatment.
The 2006 Nature paper used a commonplace lab technique called Western blot to separate out different proteins from a mixture. A bit of the mixture is placed on a flat layer of absorptive material and subjected to an electrical field. The heavier proteins move more slowly than the lighter ones. The resulting array is exposed to an antibody-based stain that allows it to be seen. The positions and sizes of the individual protein spots are then analyzed.
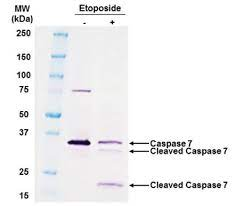
But now, a whistleblower has reported evidence that some of the Western blot images in the publication and many others from the same lab were placed where they didn’t belong, citing faint lines between blots that could result from cutting-and-pasting. There was also an instance of two blots with identical size and shape, something with a likelihood approaching zero absent a copy-and-paste operation. The journal Science hired two scientists unconnected to the Minnesota team to take a look. They confirmed that deliberate falsification is highly likely, though there’s no smoking gun, which would require access to the original Western blot images or to the original data readouts. Nor, so far, has there been a confession.
Meanwhile, what’s the upshot? For the AD field, it means that the treatment trials of anti-A-beta drugs were based on much less laboratory evidence than was thought, possibly explaining why they all failed. (Aducanumab, the antibody approved in 2021 by the FDA, targets A-beta, but its clinical benefit is highly controversial, Medicare refuses to cover the treatment, and most neurologists opt not to prescribe it.) That means that by default, anti-AD treatments addressing tau, the other protein aggregating in AD, deserve more attention.
Some experts have questioned the importance of A-beta in AD for decades, but only in the last 15 years or so has AD research into tau as the alternative received serious support. In PSP, tau is the only protein that consistently aggregates and there’s no evidence of A-beta misbehavior at all. PSP is therefore considered by many scientists to be a good test bed for anti-tau treatments for AD. That’s why I think that if these new doubts about A-beta in AD direct attention to tau, an intensification of tau-based PSP research could result, and that could, by extension, benefit AD as well.
While two anti-tau antibodies have failed to slow the progression of PSP in clinical trials, there are many other ways to address tau in PSP, including one trial currently recruiting and at least two more set to start in the next year.
So, let’s hope that this week’s revelation gives PSP research a boost and AD research a long-overdue redirection.
Here’s a detailed editorial in Science explaining all of this (without my own speculation about the possible benefit for PSP research). But it’s behind the journal’s paywall and I didn’t want to post the pdf I have access to through my university. That would be another form of dishonesty.