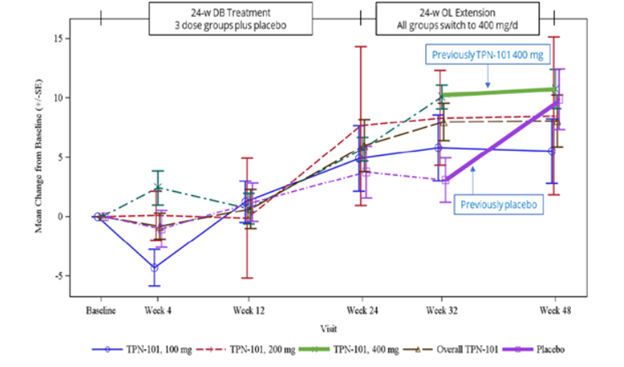
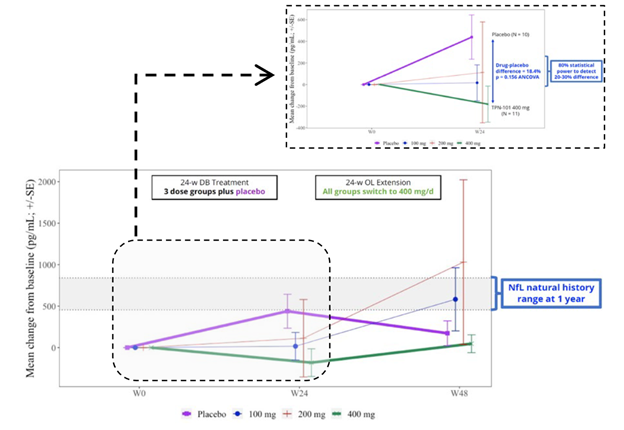
I could use your input right now. (Actually, I could always use your input, but only occasionally do I ever specifically ask for it.)
A few days ago, I attended a two-day conference in Washington, DC on the tau protein sponsored jointly by the Alzheimer’s Association, The Rainwater Foundation and CurePSP. A talk on clinical trials in the non-Alzheimer tau disorders mentioned the well-known difficulties in recruiting adequate numbers of patients with rare conditions like PSP. In the Q/A, I asked if there’s a realistic possibility of reducing the number of patients required for a trial by using a new approach called “personalized endpoints.” Afterwards, the editor of a journal introduced himself and asked me to write a review article/opinion piece on that issue. I said OK, but now I could use your help.
Here’s the background to my question at the conference, though many of you already know what’s in the next two paragraphs:
The typical Phase II or III clinical trial divides the patients into active treatment and placebo groups. Trials of chronic, progressive disorders like PSP measure the signs and symptoms for each patient at “baseline,” i.e., the first visit after the screening visit, using a battery of scales and tests. One of those, which for PSP, almost always the PSP Rating Scale, is deemed the “primary outcome measure.” Other measures of the drug’s effect are called “secondary” outcomes and still others under evaluation for future use are called “exploratory” outcomes.
At the end of the double-blind period, typically one year for PSP, the battery of outcome measures is repeated, many of them having been repeated at interim visits as well. Then, for each treatment group, disease progression is measured as the rate of progression (i.e., PSPRS points per year) from baseline to endpoint. If that difference is less, on average, for the active group than for the placebo group, and is large enough that the likelihood of having occurred by chance is less than 5 percent, then the result is deemed “statistically significant.” If that result is reinforced by similar results in at least some of the secondary outcome measures and if the side effects are justified by the efficacy given the disorder’s severity and availability of other treatments, the drug will then be considered for approval by government regulators.
So what’s the problem?
First of all, “statistically significant” is not the same as “clinically significant.” That means that a result too small to make a difference to the individual patient can, because of a study’s large size, reach statistical significance. The FDA knows this, of course, and relies on secondary outcome measures to verify clinical significance. But the secondary measures may under-perform statistically, or may measure only a single aspect of the disease such as cognition or balance, or may lie far from the patient’s lived experience, as do, for example, an MRI or a blood test.
Secondly, averaging the entire active drug group and the entire placebo group is a very coarse measure. That means that demonstrating a given treatment effect with statistical significance requires large numbers of patients and/or a longer study. Both issues mean more expense for drug companies, which means that fewer drugs will get tested, the trials will be longer, and effective drugs may appear to be ineffective (called a false-negative result or Type 2 error). None of those things is in anyone’s best interest.
So what’s the solution?
The PSP Rating Scale is far from perfect and various improvements have been published. While each improves upon the original in some way, none is more sensitive to change. The only outcome measure confirmed to be more sensitive to change than the PSPRS is the MRI-based measurement of brain atrophy, and that’s too far removed from actual symptoms and disability. So, we need new study designs that can squeeze more information out of fewer patients.
A more sensitive way to assess a drug’s benefit uses “personalized outcomes.” That’s where each patient being enrolled is assigned an expected endpoint PSP Rating Scale score based on how much they are likely to progress over the following year according to published research. Relevant baseline data includes things like age, sex, progression since onset, baseline PSPRS score and subsets thereof, certain MRI abnormalities, and levels of certain chemical markers in the spinal fluid or blood. At the end of the double-blind period, the active drug and placebo groups are compared with regard to how many patients did better than their own pre-defined expectation.
But how much better is “better”? Enter a concept called “minimal clinically important difference.” That’s exactly what it says: The smallest change in a test score that corresponds to a change that makes a difference to the patient or family. The trick is how to obtain this information in some sort of reliable, standardized way. For PSP, the only attempt to do this to my knowledge was published in 2016 by Dr. Sarah Hewer and colleagues, mostly from Alfred Hospital in Melbourne, Australia. They mined data from a completed, negative PSP trial that used a secondary outcome measure widely employed in clinical research, the “Clinical Global Impression of Change” scale (CGI-c). This simple, seven-point scale asks the study neurologist to decide if overall, compared to baseline, the patient is “very much,” “much,” or “minimally” improved; or unchanged; or “minimally, much, or very much” worse. Hewer et al, calculated the average degree of PSPRS worsening for patients rated by the CGI-c as “minimally worse” over the course of the year of the trial. (Of course, the CGI-c uses the neurologist’s opinion, not the patient’s or family’s, but hopefully, the neurologist relied on their input. I certainly always did whenever I completed a CGI-c.)
The minimal clinically important difference in the 100-point PSPRS turned out to be 5.7 points, with a 95% confidence interval (the range encompassing 95% of the patients) of 4.83–6.51. Now, 5.7 points on the PSPRS represents about six months’ decline for the average patient with PSP-Richardson’s syndrome, with most of the other subtypes declining a little more slowly.
Finally, here’s my question for you:
As the seven steps in the CGI-c may be too coarse or too fine, is a six-month decline really the “minimal” worsening that’s important to you? Or would a smaller or larger decline be the least you’d consider important? You or a helper can judge this in terms of your overall comfort, or your ability to perform daily activities, or a combination of the two. Note that I’m asking how many months’-worth of decline is important, not merely noticeable, which I assume would be less.
Please respond in the comments feature or by email: ligolbe@gmail.com. Thank you!