Three more brief items:
1
Another case of paraneoplastic PSP has been reported, making 7 in the world’s literature. This was a 62-year old woman with ductal breast cancer whose PSP-like features evolved over only 4 months to an advanced state of disability. The brain MRI and spinal fluid were normal. None of the known paraneoplastic antibodies were present in her blood. A PET scan using fluorodopa was negative, a result expected with non-degenerative forms of parkinsonism. Treatment with steroids and other immunomodulatory agents helped dramatically, restoring an independent gait and a “good quality of life” until the breast cancer became metastatic. This case was reported in the Annals of Indian Academy of Neurology by Dr. Ajith Cherian and colleagues. The take-home: When any neurodegenerative disease, including a PSP-like syndrome, evolves rapidly to disability over a period of months rather than years, a strong possibility is a paraneoplastic syndrome. That’s where the immune system’s fight against a tumor produces antibodies that also attack normal components of other organs such as the brain. Not only may the neurological symptoms improve with immunoregulatory treatment, but if those symptoms appear before the tumor has been suspected, the workup could include a thorough search for a small tumor, removal of which could be lifesaving. (Note: The information from the PET scan could have been obtained nearly as well from a dopamine transporter scan, which is far more widely available and is covered by insurance.)
2
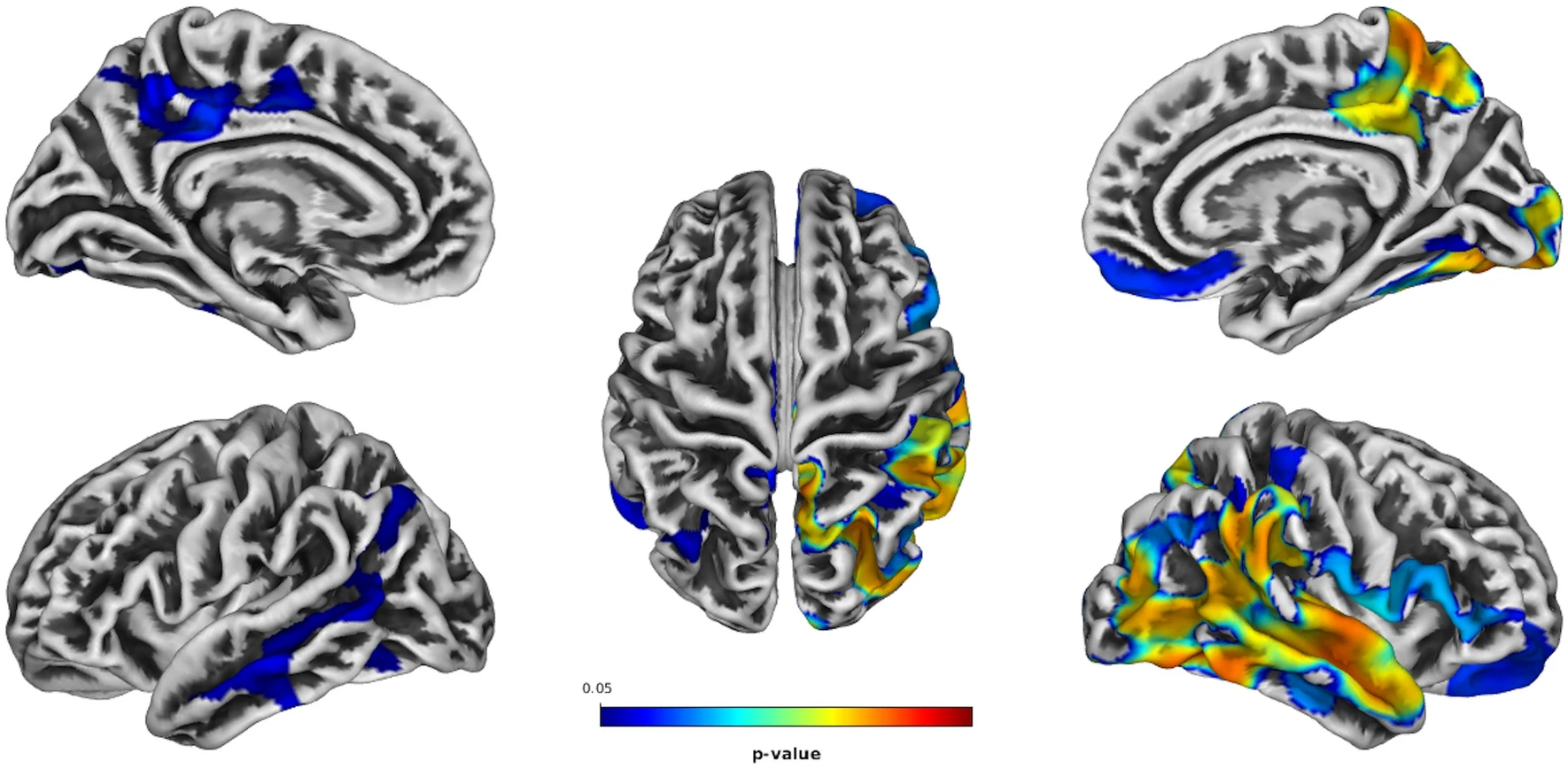
Depression occurs in about 60% of people with PSP, according to a published review. Now, an article in Journal of Neurology reports MRI scans in 40 patients with PSP, comparing the 21 with depression to the 19 without. The research group works at the University of Bari, in Tricase, at the tip of the heel of Italy’s boot, and was led by Dr. Daniele Urso, a well-published expert in neuroimaging of neurodegenerative disease. In patients with PSP and depression, they found more thinning of the cerebral cortex in the temporal, parietal and occipital lobes, moderately worse on the right than the left. (In the linked image, the colored areas are those where atrophy was greater in patients with depression than in patients without. The left cerebral hemisphere is shown on the left and the right on the right. The upper two images are the medial (inner) surface and the lower two the lateral surface.) This confirms once again that depression is part of the disease process in PSP and not (or not only) a normal psychological reaction to the overall disability. The right/left asymmetry shows that it wasn’t just that the overall PSP was more severe in the patients with depression, as the cortical atrophy of PSP is generally symmetric.
3
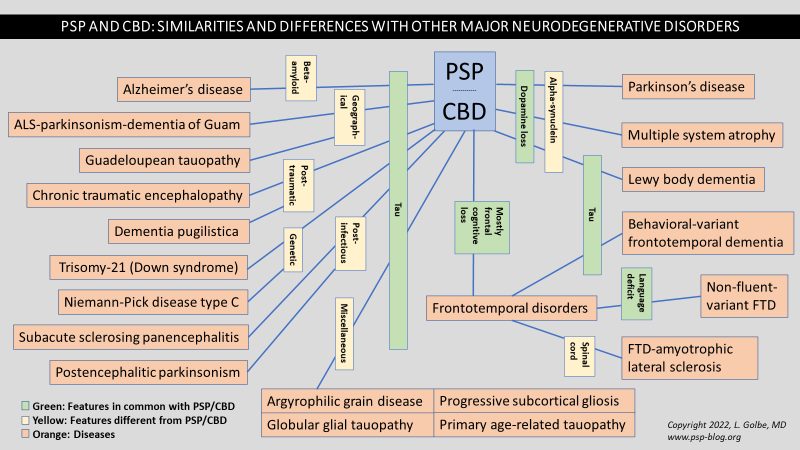
A few years ago, CurePSP created a network of 25 academic centers in the US and Canada with special expertise in care of patients with PSP and CBD, calling it the “CurePSP Centers of Care.” I’ve been one of its organizers and leaders. Last year, we published a “best-practices” article on medical management of the disorders. This year, we have expanded to 28 centers, including 2 in Canada. To keep things moving and accountable, we have doubled the size of our Steering Committee to include 4 Center directors and 4 representatives of the CurePSP Board of Directors and staff. For the first time, we’ve drafted a list of projects designed to improve clinical care and to provide clinicians of many kinds with education and awareness of PSP and CBD (and for some centers, MSA as well). Also for the first time, CurePSP is providing the Centers some modest financial support for expenses related to delivering first-rate clinical care. I’ll let you know how it’s going.


{kind=link}