You may recall a post from last week lamenting the state of diagnostic markers for PSP. But now I’m happy to report that things are starting to look up.
A paper in the current issue of Movement Disorders is from a group at Fudan University in Shanghai led by Dr. Ling Li. Two of the 17 authors work at Taiwan-based Aprinoia Therapeutics. Last on the author list is the “Progressive Supranuclear Palsy Neuroimage Initiative” (not to be confused with the 4-R Tau Neuroimaging Initiative based at UCSF under Adam Boxer). I don’t know if the PSPNI is an academically-based research group or a consortium created by Aprinoia. I’ll try to find out. In any case, Aprinoia is developing a PET tau ligand called [18F]-APN-1607, formerly known as [18F]-PM-PBB3.
First, a little background:
What’s a “PET ligand”? What’s “PET”? Positron emission tomography is a way of mapping the locations of a specific compound (called the “target”, typically a protein of some sort) in the body. First, a compound (the “ligand”) that can bind to the target, and hopefully only to that target, is formulated, and that’s the hard part scientifically. Then the ligand is attached to an atom that emits radiation, specifically positrons, for a time that’s short enough to avoid poisoning the patient or the environment. The most common positron-emitting atom is fluorine-18, but carbon-11 is another common one you’ll see. The resulting compound is injected intravenously into the patient. In about an hour or so, the ligand has bound to its target molecule. After a positron has traveled about a millimeter, it has lost enough energy that when it next hits an electron, the two annihilate each other, emitting two photons (in this case also called gamma particles) in opposite directions. The patient is precisely positioned next to a type of camera that can detect these, and when it detects two photons at exactly the same time, it calculates their common point of origin and puts a dot on its software map accordingly. The result is a series of 2-dimensional slices showing the locations of the positron emitter with its ligand. PET images are initially just shades of gray but for ease of eyeball interpretation are typically displayed in an arbitrarily chosen array of colors, with the “cool” blue colors signifying low ligand uptake and “hot” reds the highest uptake.
Although the FDA approved Tauvid (flortaucipir; [18F]-AV-1451; [18F]T807) in May 2020 as a tau-directed PET ligand for Alzheimer’s, neither that compound nor several other candidates have proven adequate in PSP. The main reasons have been that the “tau burden” in PSP is only 1% of that in AD, which makes the PET signal insufficiently distinguishable from the normal brain’s background. Also, PET in general has a much lower spatial resolution than MRI or even CT, so the small size of PSP’s specific areas of involvement makes it hard for PET to distinguish PSP from other disorders. Another issue has been non-specific binding. That is, some candidate tau PET ligands bind less to tau than to other compounds that tend to occur in the same set of brain cells but may not be affected much in PSP. A good example has been [18F-THK-5351, which distinguishes PSP from healthy people, but was found to bind mostly to monoamine oxidase B, an enzyme important in dopamine metabolism.
Another ligand,[18F]-PI-2620, has avoided that pitfall and distinguishes PSP from healthy controls. But it has not yet been shown to distinguish PSP from other atypical parkinsonisms, though adequate studies of that question have not been published. Nor has [18F]-PI-2620 been tested in patients with early PSP, where there is greatest need for a diagnostic marker – the average PSPRS score of the patients in the one published diagnostic study was 38 (0 normal, 100 worst possible), by which time PSP is usually easily diagnosable at the “bedside.” (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7341407/) Nor has that ligand been tested for its ability to distingish PSP-RS from other subtypes or to track disease progression over time.
This week’s development
The news flash is that [18F]-APN-1607, has leapt ahead of [18F]-PI-2620, at least for now. (Not that we shouldn’t have multiple tau PET ligands for PSP with slightly different properties for different clinical situations – that would be great!) The paper of Li et al included 20 patients with PSP (a lot for an early-phase PET study), of whom 16 had probable PSP-Richardson syndrome, 2 had PSP-parkinsonism, 1 had PSP-progressive gait freezing and 1 had “suggestive of” PSP. Their average PSP Rating Scale score was 31.6, which is toward the milder end of the range typical of PSP drug trials and milder than the patients in the l[18F]-PI-2620 trial. There were also 7 with MSA-parkinsonism, 10 with Parkinson’s disease (both of which are alpha-synucleinopathies, not tauopathies) and 13 healthy controls. The results were corrected for any effects of age, sex, or disease duration and for multiple comparisons.
The study found that [18F]-APN-1607 PET shows major differences between PSP and healthy controls in 12 brain regions known from autopsy studies to be affected most in PSP. The same could be said for the comparisons of PSP with Parkinson’s or MSA-P, although when only the putamen (part of the basal ganglia) was considered, 4 of the 7 patients with MSA-P had as much binding as those with PSP. So the authors combined the measurements from the substantia nigra (part of the midbrain, which is part of the brainstem) with those of the putamen, achieving much better separation. Still it was far from perfect: The standard measure of diagnostic accuracy at an individual patient level, as opposed to merely comparing two groups’ average measurements, is the area under the receiver operating curve (AUC). That statistic, where perfect is 1.0 and useless is 0.5, takes into account both sensitivity and specificity. The AUC based on [18F]-PI-2620 uptake in putamen and midbrain for PSP vs the synucleinopathies was 0.811 and for PSP vs. controls, 0.909. Good but not great.
When they homed in on the subthalamic nucleus, a tiny area that may be where PSP starts in the brain, the AUC was an excellent 0.935 (0.975 for MSA-P and 0.908 for PD). But that nucleus is so small relative to the spatial resolution of PET that it could be a problem to train large numbers of radiologists and technicians to measure it in the real world using real-world hardware and software.
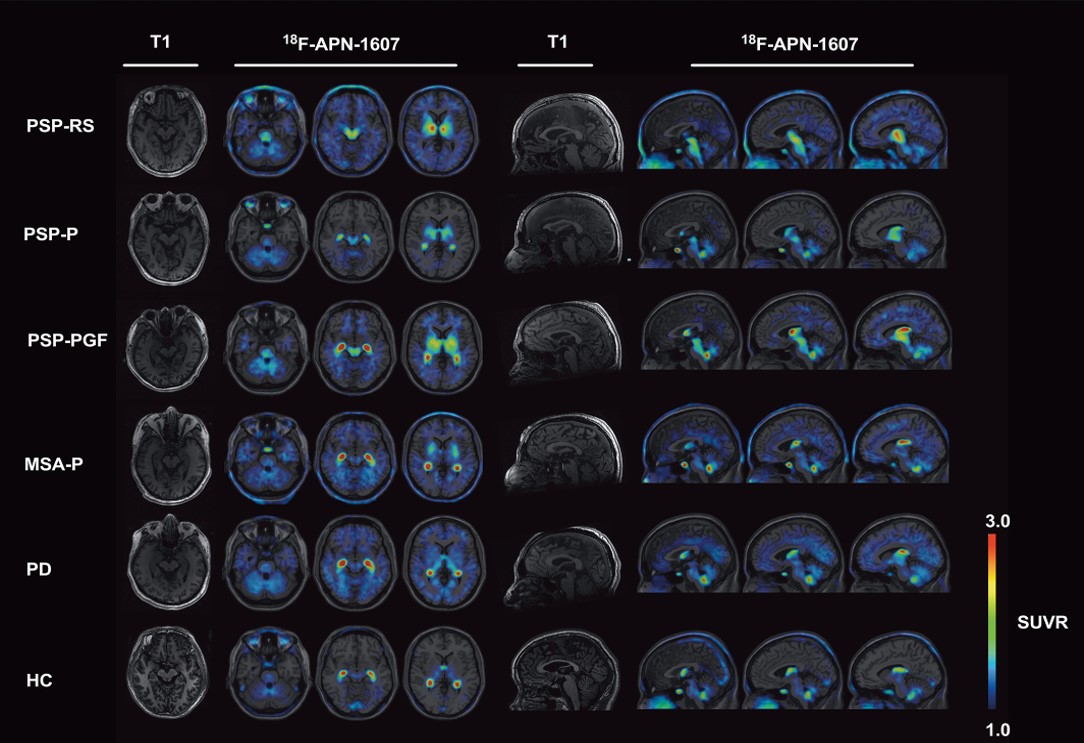
In the figure above, the first and fifth columns are the MRI images used as templates on which the PET images (the colored areas in the other columns) are superimposed. The group of images on the left are axial images through the planes of (from left to right) the pons, midbrain and putamen. On the right are sagittal images through planes a bit left of midline, midline and a bit right of midline. Each row is one patient with the condition listed at the far left. (HC means healthy control.) Note that all three subtypes of PSP show strong uptake of the tracer in the putamen and midbrain and none of the other patients shows this combination. The brain area with the greatest difference between PSP and non-PSP, the subthalamic nucleus, is too small to appear to the naked eye as a clear and separate dot in these images.
Flies in the ointment
A major pitfall for [18F]-PI-2620 is its sensitivity to light, which renders it inactive. A solution to this problem would require not only opaque containers, but also opaque IV tubing. This can be achieved by wrapping transparent tubing in foil, a standard procedure in hospitals for other photosensitive drugs, but one with obvious drawbacks.
The study of Ling et al did show, for several brain regions, a weak correlation of PSPRS score with [18F]-PI-2620 uptake. The association was best for the raphe nuclei, an area of the pons (in the brainstem) with widespread connections that use serotonin as their neurotransmitter and are most closely associated with control of sleep. Weaker, but still statistically significant associations were found also for 5 other areas. Another selling point for [18F]-PI-2620 is that the PET signal did not correlate with the subject’s age, suggesting that the uptake is related to the severity of the illness and not some effect of aging in the context of illness. However, the duration of illness did not correlate with [18F]-PI-2620 uptake, suggesting that this technique might not be able to document PSP progression or its slowing in response to treatment in a drug trial.
Another issue left untouched by the new publication is whether [18F]-PI-2620 can distinguish PSP from CBD. That would require subjecting patients with corticobasal syndrome (CBS) to amyloid scanning to rule out Alzheimer’s disease as the cause of their CBS, leaving a tauopathy as the most likely, but not the only, explanation. Nor were non-Richardson PSP subtypes evaluated, other than in those 2 patients with PSP-P.
A possible flaw in the methodology is the relatively slow progression of disability in this group of patients (on average, 0.70 PSPRS points per month, compared with about 0.92 in other studies), suggesting some sort of atypicality (or a difference of definitions of the date of onset). Another is that the PET measurements were obtained at one point in time, which may not have been the best point given the rate of brain uptake and metabolic breakdown of the [18F]-PI-2620. Using a rate of uptake over time rather than an absolute maximum would have been preferable and is the current state of the art.
Ling et al emphasize that their study is only the beginning of the clinical evaluation of [18F]-PI-2620 in PSP. Future studies should include larger numbers of patients, more non-Richardson types, CBD, and a repeat scan in each patient after 6 months or more in order to assess the ability of the technique to document disease progression in individuals.
But it’s progress!
