My post from two days ago was about a 2024 paper that had just won an award from the Alzheimer’s Association. It found and confirmed some subtle genetic risk factors for PSP. One of them has to do with the part of the immune system called the complement cascade because it complements the role of antibodies.
The finding on the complement-related gene prompted me to update my theory of the cause and pathogenesis (the subsequent sequence of events in the brain) of PSP. That includes exposure to metals, at least in some cases.
In response to that, a commenter asked which specific metals I had in mind. I responded directly in the comments section, but I thought it would make a good post for today:
—
We don’t know which specific metals might be involved in the cause of PSP, or how important they would be relative to other causes we don’t yet understand
The formal case-control surveys implicating metals asked about metals in general.Besides, that association was weak and lost statistical significance after other exposures were accounted for as confounders.
US Army veterans who developed PSP years later were more likely to have frequently fired weapons during their service than other Army veterans without PSP. So that incriminates lead, but gunfire undoubtedly aerosolizes other metals as well. My quick search reveals that gunfire can aerosolize aluminum, antimony, barium, cadmium, chromium, copper, iron, lead, manganese, nickel, tin, tungsten and zinc.
The PSP cluster in a couple of adjacent industrial towns in France suggests chromium, but that’s only the most important of the many metals contaminating that environment. Others with circumstantial associations are arsenic, copper and nickel, but others must exist as well.
The metals that caused PSP-susceptible cultured neurons to develop tauopathy in a 2020 lab study were chromium and nickel, but other metals weren’t tested because of insufficient lab capability at that time.
So that’s it, and it’s not much. It would be great if the existing genetic risk data could be analyzed for genes involved in metals detoxification. They might have fallen below the threshold of statistical significance for a genome-wide study, but a much more focused gene marker study might be able to show an association with the disease.
It could also be fruitful to analyze available autopsied brain tissue from the French cluster for metals content, comparing it to controls without PSP from the same contaminated area and elsewhere.
Also, let’s not forget that it might take certain combinations of metals, or of metals with other toxins, to increase one’s PSP risk. That could explain why there’s only one known geographical cluster of PSP — that area in France was multiply contaminated.
What’s really fun about blogging is that I can express scientific or medical opinions without having to get past experts like peer reviewers, journal editors or conference organizers. (Hence most of the evil trash on the Internet.)
But a frequent, insightful commenter called “mauraelizabeth3” asked this question after reading my last post about a new genetic finding in PSP incriminating to the myelin-producing cells, the oligodendrocytes, as a major possible starting point for PSP.
Is it known how these findings relate to (or perhaps result in) the hyperphosphorylated tau protein that aggregates in the brains of PSP patients?
I’ll respond to that excellent question with my own current theory of the etiology and pathogenesis of PSP. It’s based on legit science, but of course, I don’t really know how much emphasis to place on each of the disparate current facts, or how many additional facts await discovery.
Dear me3:
That’s really the question, isn’t it! My own formulation at this point is that the loss of myelin from those multiple gene variants is a sideshow that impairs neurological function but isn’t actually part of the cause of PSP. Instead . . .
I’d propose that the first abnormal event is some inherited or de novo mutations or epigenetic alterations in the MAPT gene (which we know do exist in PSP) changes the structure or the post-translational modifications of tau in a way that stimulates its hyperphosphorylation as a reaction designed to facilitate its degradation. Hyperphosphorylation tau might also be the result of some sort of toxin exposure, with metals currently the leading contender.
Then, hyperphosphorylated tau falls off the microtubules and is free to do mischief all over the cell. Maybe the first (or only) thing it does is to make the genomic DNA in the nucleus lose some of its protection against inappropriate transcription into RNA. It’s been shown that such inappropriate transcription allows retrotransposons to be transcribed. (Those are pieces of DNA implanted there by viruses millions of years ago. They have reproduced themselves to other parts of the genome and now account for about 40% of our DNA.)
The RNA so produced is recognized by the immune system as viral. An immune response ensues, which attacks and degrades a lot of RNA and innocent bystanders. The resulting molecular garbage is a major challenge for the cell’s regular garbage disposal, the ubiquitin-proteasome system and the autophagy/lysosomal system.
Meanwhile, all that hyperphosphorylated tau is misfolding and then aggregating with itself. This does take place under normal, healthy conditions to some extent, and the disposal systems can handle that. But now, with the garbage from the inflammation and the unusual amount of aggregated tau to degrade, the garbage disposal is overwhelmed. This allows all sorts of normal toxic garbage to accumulate, and that’s eventually fatal to the cell.
This whole process starts in the astrocytes or oligodendrocytes and spreads to the neurons courtesy of the microglia (the brain’s immune cells), synaptic connections and direct contact.
Keep in mind that I’m a clinician with little laboratory experience beyond delivering fluid samples from my patients to my smarter colleagues with the pocket protectors. But I try to keep up with the latest in all aspects of PSP, and there’s lab support for all of the assertions in my hypothesis. It’s just that putting those facts together is tricky, like cracking a complicated criminal plot. I hope that my hypothesis at least illustrates that we’re starting to get more of a handle on the pathogenesis of PSP.
Yesterday’s post was about the clinical heterogeneity of PSP and how it prompts a theory about the cause(s) of the disease. A couple of hours after I hit “send” I saw a new paper that indirectly supports my idea.
As you probably know, PSP comes in ten known subtypes. The original type, first described in detail in 1963, is called PSP-Richardson syndrome and accounts for about half of all PSP. The other nine have been described since 2005. The new paper reports five subtypes among PSP-Richardson syndrome itself.
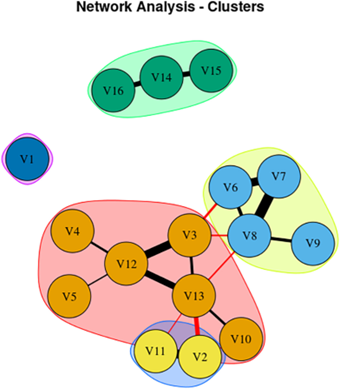
The study is from Dr. Mahesh Kumar, a post-doc at the Mayo Clinic, with Dr. Keith Josephs as senior author. They performed statistical tests called “network analysis” and “cluster analysis” on their 118 patients with PSP-Richardson. The five PSP-Richardson “sub-sub-types” emphasize, respectively, tremor; light sensitivity; reduced eye movement (i.e., supranuclear gaze palsy); cognitive loss and slowness/stiffness.
These are not just points on a continuous spectrum. Rather, in each of the five PSP-Richardson sub-sub-types, a group of features and their severities occurs together in individuals in a combination that would not be expected by random combination based their respective frequencies in the total PSP-RS population. For example, people with worse slowness/stiffness tended to have milder eye movement problems and worse cognition than chance would dictate.
Here’s a graphical representation of the results. The features represented by the circles in each group interact with one another in a mutually reinforcing (the black bars) or interfering (the red bars) way. The thickness of the bars represents the strength of the interaction. An explanation in the researchers’ own words follows:
From Kumar et al. Mov Dis Clin Prac 2025 Network Analysis showing 16 signs/symptoms and their associations. Each node in figure represents symptom/sign, Black edges represent positive connection, and red edges represent negative connection; thicker edges represent stronger association. V1, Sensitivity to bright light; V2, MoCA (Cognition Score); V3, Neck Rigidity; V4, Urinary Incontinence; V5, Emotional Incontinence; V6, Upward ocular movement dysfunction; V7, Downward ocular movement dysfunction; V8, Horizontal ocular movement dysfunction; V9, Eye lid dysfunction; V10, Limb apraxia; V11, FAB (Executive Score); V12, Gait dysfunction; V13, Bradykinesia; V14, Postural tremor; V15, Kinetic tremor; V16, Rest tremor.
All this begs the question as to the basis of the specific groups of signs and symptoms. The answer will probably apply as well to the ten PSP subtypes as to the five PSP-Richardson sub-sub-types. It probably has to do with the specific combination of PSP’s menu of causative factors at work in the individual. As I pointed out in my last post, there are 14 known gene variants contributing to PSP risk and that number is growing. Exposure to toxic metals may also be a factor and those exposures could come at different times of life and in various durations, intensities and combinations. The number of genetic/toxic combinations of these factors sufficient to cause PSP would be astronomical, and the likeliest combinations might account for the likeliest PSP subtypes and sub-sub-types.
Then throw in the stochastic factors, meaning random throws of the dice. I’ll get to that in a future post.
There are now ten different variants, or “phenotypes,” of PSP. The most common, PSP-Richardson syndrome, accounts for about half of those affected, and the next most common, PSP-Parkinsonism, accounts for about a third. All ten variants share the same kinds of neurofibrillary tangles, tufted astrocytes and all the other microscopic features, but their specific locations in the brain differ in emphasis. In fact, the ten have been classified into three groups: cortical PSP, subcortical PSP and PSP-RS, the last being a sort of hybrid. The differences among the ten exist only for about the first half of the disease course. After that, they all merge into what looks like PSP-RS.
What explains this (slight) diversity of anatomic predilection? We won’t know that until we know the cause of PSP, but I’ve got my theory, which I’ll tell you about after you consider this:
Consideration number two:
In 2021 I posted something about a geographical cluster of 101 people with PSP in a group of towns in northern France, which is 12 times the number expected based on surveys elsewhere. The most likely cause is the intense environmental contamination with metals dumped there by an ore processing plant. Fortunately, there have been no new cases of PSP in that area since 2016, possibly thanks to the mitigation measures taken by the local authorities starting in 2011.
In a 2015 journal publication, I did some calculations comparing the neurological features in the 92 people with PSP in the cluster at that point to those of people with “sporadic” PSP.
I found only two differences: In the cluster, the ratio of PSP-Richardson syndrome to PSP-Parkinsonism was about even, while in sporadic PSP it’s about 3:2; and the average age of symptom onset was 74.3, about a decade older than in sporadic PSP. (The area was not at all a retirement community; its age frequency structure closely resembled those of other ordinary communities in the industrialized world.) The molecular assays we performed showed no differences.
My theory:
The experts agree that the cause of most of the common neurodegenerative diseases is a genetic predisposition together with an environmental exposure. For PSP, we presently know of 14 genes, each of which has a variant in a certain percentage of the population conferring slightly elevated risk of PSP. But we don’t know how many, or what combination of those 14 are needed to set the stage for the environmental toxin. For all we know, different toxins need different numbers or combinations of PSP genetic risk factors to exert their toxicity. The only confirmed environmental risk factors for PSP are metals, but of unspecified kinds. The only other well-confirmed non-genetic PSP risk is a tendency to lesser educational attainment, which I feel is likely to act by exposing people to toxins related to manual occupations or to industrial installations or waste sites near their homes.
So, here’s how I tie all this together:
The ten PSP variants as well as the diversity of onset ages within each variant could be determined by one’s own set of PSP risk genes and by which of the possible PSP-related metals (or yet-undiscovered kinds of toxins) they encountered. The two differences between the French cluster and everyone else with PSP could be the particular types or combination of metals to which the people were exposed. That means that at its root, the cause of PSP could be an array of slightly different abnormalities at the most basic molecular level. Those differences could take the form of slightly different tau protein abnormalities across different individuals. As has already been shown, each member of the array of abnormal forms of tau (called tau “strains”) might have a predilection for a different brain area or brain cell type. That anatomic predilection would dictate the specific array of symptoms initially experienced by that individual, and that array could be different when your tau was damaged by the metals at the French site than by whatever damages tau in PSP elsewhere.
How to prove this theory:
This would take a lot of difficult research to prove, but I made a start back in 2020 with the publication of some lab experiments I suggested to a group of lab scientists at UCSF led by Dr. Aimee Kao.
She and her colleagues took stem cells from a tiny skin biopsy of a person with ordinary PSP who carried one of the known PSP risk genes. They converted the stem cells into brain cells and divided the resulting colony in two: In one half, they used the now-famous gene editing technique called CRISPR to return the variant to its normal state and left the other half with its PSP risk variant. They added chromium or nickel (the two most likely culprits at the French cluster site) to both sets of cells and found that the corrected set suffered much less damage. Furthermore, the damage involved tau aggregation and insufficient disposal of abnormal tau, just as in PSP itself.
So, now that we know 14 PSP risk genes, lab researchers could experiment with stem cells harboring different combinations thereof, along with exposure to different metals. Then, once a few such gene/metal combinations have been identified as most able to cause “PSP” in stem cells, the underlying molecular abnormalities could be worked out, drug targets identified, and drugs designed, tested, approved and prescribed.
Since 2014, I’ve been trying to find the cause of a geographical cluster of PSP in northern France. It’s the only documented PSP cluster known. The problem was difficult enough, but now the cluster has mysteriously disappeared.
The clinical aspects of the cluster are detailed in this 2015 paper. Here’s the executive summary: In 2005, Dominique Caparros-Lefebvre, MD, a geriatric neurologist with experience in PSP research, arrived at her new practice position in Wattrelos, France, an industrial suburb of Lille. By 2007, she started to notice more PSP than expected and developed an excellent database. She diagnosed 100 patients over the next decade. In 2013, she invited me to help her find the cause, as I had had some experience in the epidemiology of PSP. I calculated the observed-to-expected incidence ratio of PSP to be 12.3 in Wattrelos and its neighbor to the south, Leers. Most clusters of chronic diseases such as cancer have ratios much lower, in the range of 1.5 or 2.0. So this was a major cluster.
PSP in the 100 patients has differed only slightly from “sporadic PSP,” with more PSP-Parkinsonism than PSP-Richardson syndrome, and an older mean onset age, 74.3. The 13 autopsied cases show typical PSP, with the expected 4R tau and the H1/H1 genetic haplotype. That work was done by a very accomplished research team at the University of Lille led by Luc Bueé, PhD and Vincent Deramecourt, MD, PhD. No other molecular genetic workup has been performed to date, but none of the affected persons were related to one another and among the patients are 7 Algerian immigrants, a strong point against a genetic origin.
Wattrelos and Leers have extensive chemical contamination, especially by metals from an ore extraction plant that operated in southern Wattrelos for most of the 20th century. Huge piles of spent chromate and phosphate ore, now covered, remain on the plant’s property, which has been converted into to a public park after mitigation efforts between 2000 and 2010. Only 2 of the 100 patients worked in the chromate/phosphate ore plant, but soil from the area adjacent to the slag heaps has been used as fill in construction and road maintenance over a wide area. Furthermore, multiple chemical-related industries such as tanning and dyeing formed the base of the town’s economy for many decades.
So, the obvious culprit has been metals. Chromium is a carcinogen but not a good candidate as a direct neurotoxin, as its most common form, hexavalent chromium, does not cross the blood brain barrier. Nor is phosphorus a good candidate, but phosphate ore often contains important levels of other metals.
In France, growing one’s own herbs and vegetables is a common practice, even in densely urban areas. Dr. Caparros suspected thyme, a widely used herb in French cooking that avidly absorbs metals from the soil. The French government’s soil data and our analysis (by my Rutgers colleague Brian Buckley, PhD) of home-grown thyme samples from Wattrelos suggested that arsenic, cadmium and nickel were the most likely possibilities.
In 2016, I recruited a team of neuroscientists led by Aimee Kao, PhD of UCSF, with skills in stem cell models of PSP and access to stem cells with PSP-related tau gene mutations. As an initial project, they created brain cells with a rare PSP risk mutation (to create a “background” vulnerability) and exposed them to chromium, cadmium and nickel. They did the same experiment with cells from the same PSP patient except that the PSP risk mutation was converted to normal using CRISPR. They found that some of the same damage seen in PSP — aggregation of tau and evidence of apoptotic (i.e., programmed cell death) in the exposed cells with the mutation. But those abnormalities are not specific for PSP. We published that in 2020 and unfortunately, I couldn’t keep that team together for follow-on projects. Equally unfortunately, the local French human research authority would not allow Dr. Caparros to perform further field work that might have pointed to a specific metal and route of exposure.
So why aren’t clusters of PSP seen in the many other places in the world where those metals contaminate the environment? My own pet theory was that toxicity from multiple metals acting in concert is needed, and no place other than Wattrelos/Leers has a combination of so many metals in one spot together with a physician able to diagnose PSP as well as Dr. Caparros. So, one of the follow-on projects might be to repeat the lab experiments with combinations of the same and other metals that are known to occur in the environment, either in Wattrelos/Leers or elsewhere, either as a result of industrial pollution or naturally occurring.
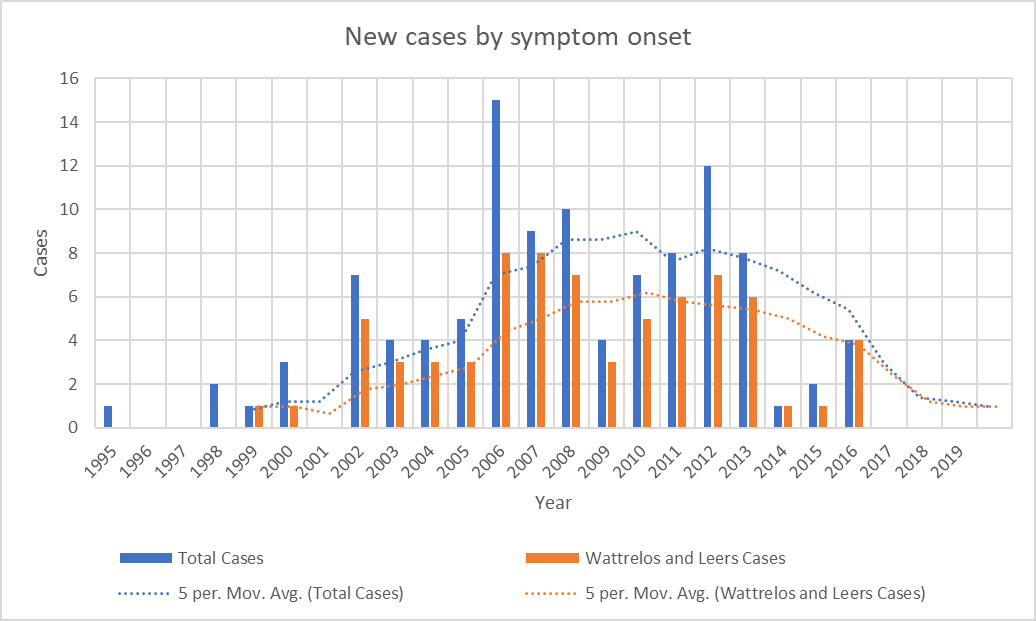
As I was starting to make plans for such a project with lab colleagues at Rutgers, it became clear that the number of patients whose onset occurred since 2013 has been declining. The most recent onset date of the 100 patients is 2016. This is not how a cluster of toxic (or genetic) cause should act. It’s possible that the mitigation efforts on the two slag heaps reduced the rate of entry of chromium and phosphate into the local environment, but the metals were pervasive in the area and presumably remain so.
—-
Graph showing the disappearance of cases with onset since 2016. Bars show the number of cases with onset in each year. Dotted lines show the average over the previous 5 years. “Total Cases” refers to all cases in Wattrelos and Leers as well as in nearby towns where patients were likely to use the Wattrelos hospital. The paucity of cases with onset before 2002 is explained by the arrival of Dr. Caparros in Wattrelos in 2005. Before that, no physician likely to have been able to diagnose PSP worked there and few patients with onset before 2002 would have survived to come to Dr. Caparros’ attention. Still, we cannot rule out the possibility that the cluster in fact started in the 1990s and its disappearance by 2017 is consistent with this possibility.
—-
The unexplained abatement of a geographical or temporal disease cluster speaks for an infectious cause. The salient example of an infection causing a temporal cluster of a neurodegenerative tauopathy is postencephalitic parkinsonism (PEP). That cluster started 2 years before and ended a decade after the great “Spanish” flu pandemic of 1918-1920 and is independent from it. PEP was a chronic, levodopa-responsive parkinsonism that affected people of any age who recovered from an encephalitis that was presumably viral, but the specific virus has not been identified. At autopsy, the brain showed neurofibrillary tangles not very different from those of PSP. The last patients with PEP died before modern molecular techniques were available, so its cause may never be known.
Could the cause of the Wattrelos/Leers cluster have been a virus? True, there seemed to have been no antecedent encephalitis, but it may have been mild, self-diagnosed as a cold or the flu, and forgotten by the time Dr. Caparros saw the patient decades later. But there need not have been any clear acute-phase symptoms at all. The virus could have set up a slow process of damage involving tau aggregation, starting with inserting its own genetic material into that of the host. Or the initial infection could have altered the patient’s immune system in a way that encouraged (or allowed) the pathology of PSP to develop. Let’s not forget that disordered immune modulation is one of the up-and-coming theories of PSP-causative factors.
If a virus contributed importantly to the cluster, could ordinary, “sporadic” PSP outside of the cluster be the result of a similar virus? Or maybe sporadic PSP is caused by the same virus without the predisposing local factor of the unusual metals exposure. Or maybe a virus infected the gut microbiome of the Wattrelos population in a way that increased PSP risk. I could go on.
We know that at least one neurodegenerative condition, sporadic Creutzfeldt-Jakob disease, is caused by an infectious agent (in this case the prion protein) without geographical or temporal clustering. The idea of a virus or prion as a cause of PSP is not new, and previous attempts to prove that hypothesis starting in the 1970s have been negative. But the technology for finding viral fingerprints has improved markedly since then.
I’ll try to get some of the research honchos I know interested in this theory and get back to you.