Some encouraging news from the AD/PD Conference held this past March in Copenhagen. NIO-752 is the anti-sense oligonucleotide drug being developed by Novartis for PSP and Alzheimer’s. It reduces the ability of the messenger RNA encoded by the tau gene to be translated into tau protein. The news is the results of a Phase I trial designed mainly to assess safety and tolerability of various dosage levels. But the trial also included measures of efficacy, just in case something dramatic appeared despite the trial’s small size. The PowerPoint slides from the presentation by Dr. Günter Höglinger of Munich can be downloaded here.
NIO-752 is a large molecule that can’t cross the blood-brain barrier, so it has to be injected directly into the spinal fluid via the same sort of needle insertion used in a diagnostic spinal tap. In this trial, 80% of the 59 subjects (45 on active drug, 14 on placebo) received injections at baseline and at months 1, 2 and 3 at ascending dosage levels. The other 20% received it at baseline and at months 3, 6 and 9. The final assessment for all subjects occurred at 12 months. The trial ran from February 2021 to October 2024.
The drug caused very little by way of important side effects: confusion and or lethargy in two of the 25 patients on the two highest dosage levels and some brain inflammation in one patient on the highest level as evidenced by elevated white blood cells in the spinal fluid. This is a very modest overall burden of adverse effects, and what’s more, the frequency of milder side effects was no different between the active drug (19 of 45) and placebo (7 of 14) groups. There’s lots more information at clinicaltrials.gov.
An important goal of Phase I trials is to demonstrate “target engagement.” That military-style term means the ability of a drug to accomplish its job in the body’s tissues regardless of whether it actually helps the person’s symptoms or long-term outcome. NIO-752 did well on that score, reducing the spinal fluid tau levels, especially at the highest dose level. (The level remained unchanged in the placebo subjects.) It also prevented any rise in levels of neurofilament light chain (NfL), while the placebo group’s NfL rose by nearly 40%. NfL is a sign of damage to axons (the long fibers emerging from brain cells) that is elevated in PSP and several other neurodegenerative diseases.
The question in your minds is, “But what about the rate of worsening of the symptoms and disabilities”? That wasn’t reported because there weren’t enough patients to perform a proper statistical analysis. If the worsening in the PSP Rating Scale had been less in the 45 participants on NIO-752 than in the 14 on placebo, that would be too few to exclude the possibility of confounders (a “Type I error” or false-positive) effect. Similarly, if this small study failed to demonstrate a benefit, Novartis would not want anyone to draw negative conclusions about the drug without a properly powered trial.
Next step: Novartis will now skip directly to a Phase III trial and just yesterday they posted details on clinicaltrials.gov. You can get a look here, but I’ll discuss that trial in a near-future blog post. The trial’s nickname is PRESERVE.
__
Full disclosure: I have consulted for Novartis on trial design in the past but not since 2023. I have no stock in the company nor any other financial interest in the success of NIO-752 or the company in general.
It’s been a year since I promised you an explanation of the role of “stochastics” in PSP and other neurodegenerative diseases. It goes to the questions I’ve heard from every patient with PSP I’ve ever treated: “Why me?”
The cause of a disease can be boiled down to two components: etiology and pathogenesis. Pathogenesis is about the processes in the body that produce symptoms and tissue loss – the topic of many of my blog posts, but not this one. Etiology is about causative factors originating outside the individual. Here’s a very generic rundown of what’s known about the etiology of PSP:
Variants in the genetic code inherited from one’s parents:
15 gene variants are known to increase the risk of PSP, though each has only a small effect and together they probably explain less than a quarter of the total population’s PSP risk.
A paper published a few days ago found that a common thread in 13 of the 15 was impairment in the microtubules, the brain cells’ internal monorail/skeletal system. But other commonalities could exist, too.
Experiences during life, such as:
Lesser educational attainment is associated with PSP risk.
Other experiences associated with other neurodegenerative diseases include minor brain injury and non-specific stress.
Toxic exposures
Rural living and well water use, each possibly via pesticide exposure.
Metals, though the specific metals and the routes of exposure remain unclear.
Foods such as sweetsop, soursop, and American paw-paw, with toxins affecting the mitochondria.
But all these together, based on my seat-of-the-pants statistics, don’t explain most of the population’s risk of developing PSP. Other than genes, experiences and toxins that have eluded detection to date, what other suspects could there be? Stochastic events.
That word basically means “random.” What specific events are happening randomly to cause PSP? A few possibilities:
Random mutations in one’s DNA occurring during cell division (called “somatic” mutations, as distinguished from “germ line” mutations from mom and dad) may fail to be corrected by the brain’s error-correction machinery.
Random errors in the encoding of RNA from normal DNA.
Random changes to the DNA other than the nucleotide sequence (the “letters” in the genetic code) itself. Such changes usually consist of small molecules attached to the DNA and are called “epigenetic” changes. They occur normally as a way to regulate gene function but can also occur inappropriately, with harmful result.
But the one I’ll put my money on is random tau protein misfolding
Here’s how that works: Normal tau has no standard pattern of folding on itself. Rather, each normal tau molecule is like a piece of overcooked spaghetti in boiling water. But occasionally, and randomly, the loops and curls of one strand happen upon an arrangement that sticks to itself. The brain does have an app for that – a sophisticated mechanism to recognize, tag, and dispose of such miscreants. But some of those abnormal folding patterns have the unfortunate ability to get nearby normal copies of tau to adopt the same abnormal folds. This process, as you’d imagine, operates as a chain reaction, with each misfolded tau molecule inducing the same change in others. The misfolded molecules tend to form stacks, like checkers with interlocking ridges. Those stacks are called fibrils and they’re toxic. Clusters of fibrils are called neurofibrillary tangles.
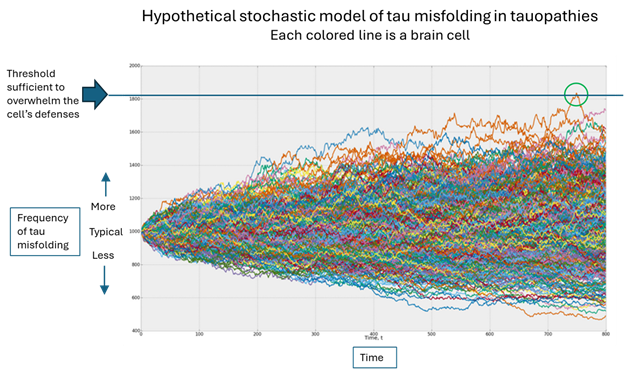
Which brings us to the original question, “Why me”? In the figure below, the horizontal axis is time, the vertical is the population of misfolded tau and each colored line is one brain cell. (The graph was designed by an investment advisory service called Artificall.com to describe the random behavior of stock prices, but the principle is similar. I’ve adapted the graph to present purposes. Ignore the tiny number labels.)
Here’s what’s happening, in my opinion: Each brain cell starts out with the same frequency of tau misfolding events, but then that frequency varies randomly. In the vast majority of cells, the resulting number of misfolded tau molecules stays within the range that the cell’s disposal system can handle. But very rarely, one cell’s load of misfolded tau molecules exceeds that limit (the green circle) and the process of templating more copies can proceed.
Once that one cell on which I’ve placed the green circle has exceeded its ability to dispose of misfolded tau molecules, it can start transmitting them to nearby cells through both synapses and direct contact without synapses. As the cell dies from the toxic effects of all those misfolded tau molecules, it will burst, allowing its misfolded tau molecules to disperse through the brain’s fluid to more distant areas, where the same process occurs.
Where does the “randomness” come in?
Notice that each colored line in the figure varies randomly, its direction of variation at any given point being independent of the movements that got it there. Sooner or later, one brain cell will, by pure chance, accumulate enough random variations in the graph’s upward direction to reach the threshold that overwhelms the cell’s defenses. (Note that an equal number of brain cells are enjoying a less-than-average frequency of tau misfolding – again randomly.)
Where do the other causative factors come in?
Without getting into the weeds, those things damage other cellular functions, perhaps in a very subtle way, but enough to impair the disposal mechanism a bit, thereby slightly lowering that horizontal blue line in the graph, which in turn increases the chances that one brain cell will see its disposal threshold exceeded.
So, what’s the takeaway?
We can’t control the laws of statistics driving the randomness. But we can look at the mechanisms of the known factors that lower the level of that blue line, identify drug targets that neutralize those actions, and design drugs (or repurpose existing molecules) to interfere with them.
So, despite all the energy I’ve put into the genetics and environmental epidemiology of PSP and Parkinson’s over the course of my career, I’ll say this: Maybe it’s time to stop looking for more little contributing causes. Instead, maybe we should devote more of our resources to designing and testing drugs that fit and influence the function of known proteins critical to the processes by which randomly misfolded tau causes damage. AI tools already exist for this purpose and are in active use.
Another approach the problem of low-impact risk factors is to look at them in combination. In theory, they could interact with one another to elevate PSP risk. But that would require either very large patient surveys or sophisticated laboratory models such as stem cells or mice with one or more PSP-related genetic mutations that are then exposed to pesticides or metals, etc.
I’ll keep you apprised before another year passes.
Today’s New York Times had a human interest story about people with a rare, genetic form of amyotrophic lateral sclerosis (ALS; Lou Gehrig disease) who are benefiting from a drug called tofersen (brand name, Qalsody). It was approved for clinical use in the US in 2023 and in Europe in 2024. The drug slows the progression of that rare form of ALS by about two-thirds, a phenomenal degree of efficacy. Today’s story was not news, just a heart-warming a review of the experiences of a few of the people benefiting.
Tofersen is a member of a drug class called “anti-sense oligonucleotides” (ASOs). If that sounds familiar, it’s because several other drugs with the same mechanism are being developed for PSP. ASO’s interfere with the ability of one’s cells to manufacture a specific protein. In the case of PSP, that protein is tau, and for ALS, it’s superoxide dismutase-1 (SOD-1). The FDA approval and the NY Times story pertain only to the 1-2% of ALS sufferers with an inherited mutation in the SOD-1 gene. However, a 30-subject, non-blindedtrial of tofersen in people with ALSwithout an SOD-1 mutation (that is, the vast majority) is in progress at Washington University in St. Louis, under the direction of Dr. Timothy Miller and colleagues, the drug’s original discoverers. That trial is scheduled to end in 2028.
As far as PSP is concerned, the ASO furthest along the pipeline is NIO-752, from Novartis.That Phase 3 trial is scheduled to start this month (May 2026) with 300 patients with non-familial PSP (as for ALS, the vast majority).
Should we expect a two-thirds slowing of progression, as in ALS with SOD-1 mutations? Probably not, for two reasons:
There’s no single mutation producing abnormal tau protein in the vast majority of people with PSP.
ASOs are large molecules – too large to cross the blood-brain barrier. So, they are injected directly into the spinal fluid using the same procedure a diagnostic spinal tap. ALS is a disease mostly of the spinal cord, which is close to the injection site and only a fraction of an inch in diameter, so tofersen can easily soak into the cord’s full thickness. PSP, on the other hand, is mostly a disease of the brain, where a drug must penetrate a longer distance and into a much larger mass of tissue. It has been shown to do so in monkeys, but our large human brains may be a different story.
Despite those caveats, I’m optimistic because even if PSP derives only half of the benefit enjoyed by this genetic form of ALS, it will be a huge advance. Scientists call this “proof of principle.” That means that the general idea has been found to make sense in a similar situation.
The list of centers slated to participate in the NIO-752 trial has not been announced, so if you’re interested, keep an eye on www.clinicaltrials.gov, www.curepsp.org or this blog. Before you volunteer, keep in mind that several other promising trials for PSP will be starting over the next few months. Check those same three sources for info on those.
(Disclosure: I’ve done consulting for Novartis, but none since 2023, and I have no financial interest in the company.)
Yesterday’s post was the first five of my top ten PSP news items of 2025. Here are the rest, again in approximate and subjective descending order of importance.
New ways of interpreting standard MRI images have gained ground as diagnostic markers for PSP. One is a test of iron content in brain cells called “quantitative susceptibility mapping” (QSM). Nine papers on that topic appeared in 2025, four in 2024 and none previously. It’s looking like combining QSM data from ordinary measurements of atrophy of PSP-related brain regions could be the ticket, as both measures come from the same test procedure, unpleasant though it may be, and they measure different things.
Positron emission tomography (PET) imaging of PSP’s type of tau (“4-repeat tau”) has made advances in 2025. This test requires intravenous injection of a “tracer” with a radioactive component that enters the brain tissue,sticks to the target molecule and is then imaged. It can distinguish PSP from non-PSP, distinguish among various PSP subtypes, and quantify the disease progression. The leading such tracer in terms of readiness for submission to the FDA is [18F]PI2620 and a distant second is [18F]APN-1607 ([18F]-PM-PBB3; Florzolotau). A tau PET tracer called Flortaucipir is on the market as a test for Alzheimer’s disease, but it performs poorly for PSP.
There’s brain inflammation in PSP, but it’s not clear whether it’s a cause or a result of the loss of brain cells, or both. Regardless, measuring the quantity and type of inflammation using blood or PET could shed light on the cause of the disease, identify new drug targets, and serve as a diagnostic marker. A good example of 2025 research on blood markers of inflammation in PSP is here and on PET imaging of inflammation is here .
We know of variants in 21 different genes, and counting, each of which subtly influences the risk of developing PSP or its age of onset. The area of the genome most important to PSP is the one that includes the gene encoding tau (called “MAPT”) on chromosome 17. The most important PSP genetic advance in 2025 was probably the discovery that some PSP risk is conferred by extra copies of a stretch of DNA, not the sequence itself. This news could inspire investigation of other places in the genome for other copy-number variants, which are much trickier to find than sequence variants. Here’s a great review of the latest in PSP genetics.
And lastly, a disappointment: a negative result of a double-blind trial of the combination of two drugs already approved for other conditions: sodium phenylbutyrate (“Buphenyl”) and taurursodeoxycholic acid (“TUDCA”). Blog post here.Sponsor’s press release here. Buphenyl protects the endoplasmic reticulum, which helps manufacture proteins, and TUDCA helps prevent brain cells from undergoing self-destruction (“apoptosis”) in response to various kinds of stressors. The pair were theorized to act synergistically. The trial’s upside is that its placebo group data can be used to provide better statistical support for future innovations in clinical trial design.
Maybe I’m streaming too many dramatic TV series these days. My October 9 post ended in a cliffhanger, teasing an “oddball” molecule that could point the way to neuroprotective treatments for PSP and other neurodegenerative diseases. It’s called “lncRNA FAM151B-DT.”
Quickly, some background. The RNA most familiar to us is messenger RNA. Its length can be anywhere from a few hundred to a few thousand base pairs (the genetic code’s “letters” for a single gene or a fragment thereof). The RNA is constructed (“transcribed”) in the cell’s nucleus from the code in DNA, then scoots out to the ribosomes, where it’s translated into a string of amino acids to build a specific protein. But only about two percent of the DNA in our genome encodes the kind of RNA for making proteins, called messenger RNA. Most of the rest, about 75 to 90 percent, encodes RNA that regulates DNA transcription or other cell functions. A little of that “non-coding RNA” is “micro-RNA,” which has only about 20 to 25 base pairs, and the rest, with over 200 base pairs, is called “long, non-coding RNA.”
Now I’ll get to the point. A research group at Washington University in St. Louis just published a paper entitled, “A novel lncRNA FAM151B-DT regulates degradation of aggregation prone proteins.” They used brain cells obtained at autopsy from people who had died with PSP, Alzheimer’s, or Parkinson’s disease. They also used skin cells from a living person with a form of frontotemporal dementia with Parkinsonism (FTDP), which is caused by a mutation in the tau gene. They transformed those (slightly) specialized skin cells into unspecialized stem cells, then transformed those into highly specialized brain cells.
The lead author of the WashU study is Arun Renganathan, PhD, a staff scientist in the Department of Psychiatry. The senior author is Celeste Karch, PhD, associate professor of psychiatry. Disclosure: Dr. Karch and I have collaborated in research in the pastand she’s a member of CurePSP’s Scientific Advisory Board, which I am honored to chair.
In each of those four disease-specific brain cell cultures, the team found FAM151B-DT reduced relative to control cells and that silencing FAM151B-DT by “knocking out” its gene increased the concentration of whichever protein was aggregating in the corresponding human disease (tau for PSP, AD and FTD-P; alpha-synuclein for PD). The mechanism was a blockage of autophagy, an important component of brain cells’ “garbage disposal” system. The researchers found that FAM151B-DT serves as a “scaffold” to allow the tau or alpha-synuclein protein and a “chaperone” molecule called HSC70 to interact with the lysosomes, a kind of bubble in the cell fluid containing protein-degrading enzymes.
A critical piece of the new research is that increasing the cells’ production of FAM151B-DT stimulated that system to dispose of excess tau or alpha-synuclein. That means that FAM151B-DT is the “rate-limiting step” in the process. As you’d imagine, this suggests that increasing the concentration or efficiency of FAM151B-DT could slow or halt progression of these diseases. All four of them.
So, how does this relate to the cliffhanger from yesterday’s post about our evolving perspective on the similarities and differences between PSP and AD? One reason to be interested in the differences between those two is that a rare disease with limited research funding like PSP could benefit from research on treatments for AD, a very common disease with much more research funding and huge commercial potential. Besides, we in the PSP community like when drug companies try out their AD drugs on PSP first – because of their common underlying cellular and biochemical similarities. The new paper from WashU has found one more very important similarity.
It’s not only PSP and AD. The new paper found FAM151B-DT just as relevant to PD and FTDP. I expect to see research soon on its relevance to others forms of FTD and to ALS, dementia with Lewy bodies, corticobasal degeneration, multiple system atrophy, and many others. Then we wouldn’t have to worry so much about making an accurate diagnosis early in the disease course– maybe one cure will fit all!
What’s really fun about blogging is that I can express scientific or medical opinions without having to get past experts like peer reviewers, journal editors or conference organizers. (Hence most of the evil trash on the Internet.)
But a frequent, insightful commenter called “mauraelizabeth3” asked this question after reading my last post about a new genetic finding in PSP incriminating to the myelin-producing cells, the oligodendrocytes, as a major possible starting point for PSP.
Is it known how these findings relate to (or perhaps result in) the hyperphosphorylated tau protein that aggregates in the brains of PSP patients?
I’ll respond to that excellent question with my own current theory of the etiology and pathogenesis of PSP. It’s based on legit science, but of course, I don’t really know how much emphasis to place on each of the disparate current facts, or how many additional facts await discovery.
Dear me3:
That’s really the question, isn’t it! My own formulation at this point is that the loss of myelin from those multiple gene variants is a sideshow that impairs neurological function but isn’t actually part of the cause of PSP. Instead . . .
I’d propose that the first abnormal event is some inherited or de novo mutations or epigenetic alterations in the MAPT gene (which we know do exist in PSP) changes the structure or the post-translational modifications of tau in a way that stimulates its hyperphosphorylation as a reaction designed to facilitate its degradation. Hyperphosphorylation tau might also be the result of some sort of toxin exposure, with metals currently the leading contender.
Then, hyperphosphorylated tau falls off the microtubules and is free to do mischief all over the cell. Maybe the first (or only) thing it does is to make the genomic DNA in the nucleus lose some of its protection against inappropriate transcription into RNA. It’s been shown that such inappropriate transcription allows retrotransposons to be transcribed. (Those are pieces of DNA implanted there by viruses millions of years ago. They have reproduced themselves to other parts of the genome and now account for about 40% of our DNA.)
The RNA so produced is recognized by the immune system as viral. An immune response ensues, which attacks and degrades a lot of RNA and innocent bystanders. The resulting molecular garbage is a major challenge for the cell’s regular garbage disposal, the ubiquitin-proteasome system and the autophagy/lysosomal system.
Meanwhile, all that hyperphosphorylated tau is misfolding and then aggregating with itself. This does take place under normal, healthy conditions to some extent, and the disposal systems can handle that. But now, with the garbage from the inflammation and the unusual amount of aggregated tau to degrade, the garbage disposal is overwhelmed. This allows all sorts of normal toxic garbage to accumulate, and that’s eventually fatal to the cell.
This whole process starts in the astrocytes or oligodendrocytes and spreads to the neurons courtesy of the microglia (the brain’s immune cells), synaptic connections and direct contact.
Keep in mind that I’m a clinician with little laboratory experience beyond delivering fluid samples from my patients to my smarter colleagues with the pocket protectors. But I try to keep up with the latest in all aspects of PSP, and there’s lab support for all of the assertions in my hypothesis. It’s just that putting those facts together is tricky, like cracking a complicated criminal plot. I hope that my hypothesis at least illustrates that we’re starting to get more of a handle on the pathogenesis of PSP.
I want to tell you about one small study that, although it needs expansion and confirmation, is exciting because it could allow a future PSP neuroprotective treatment to be prescribed before having to wait for symptoms to appear.
You’ve probably heard of the protein “alpha-synuclein” (pronounced “suh-NOO-klee-in”). It has a long list of normal functions in our brain cells, but in its various misfolded forms, is the major component of abnormal protein aggregates of Parkinson’s disease, dementia with Lewy bodies and multiple system atrophy. Now, a blood test for alpha-synuclein might actually provide a way to diagnose PSP despite the fact that tau, not alpha-synuclein, is PSP’s abnormally aggregating protein.
The new paper in the journal Biomedicines by researchers at the University of Catanzaro in Italy, have found the concentration of alpha-synuclein to be slightly greater in the red blood cells of people with PSP than in healthy people or those with PD. The graph below from the journal article (with my explanatory notes and arrows) compares results from the eight people with PSP to 19 with PD and 18 healthy control participants. The vertical axis is alpha-synuclein concentration expressed in nanograms of alpha-synuclein per milligram of red blood cells.
One paragraph of technical background on alpha-synuclein: While alpha-synuclein does most of its work in brain cells, helping in neurotransmitter release and protect against mis-application of the cell’s “suicide” program (called “apoptosis”), it’s also abundant in red blood cells. In fact, it’s the second-most-abundant protein in red cells after, of course, hemoglobin. The job of alpha-synuclein there is to help to stabilize their red cells’ outer membranes and to help in the process of removing the nucleus from the red cells’ precursor cells in the bone marrow. Nucleus removal makes more room for hemoglobin and more important, allows the cells to deform more easily as they pass through capillaries. That deformation provides a signal to the hemoglobin to release their oxygen to the tissue.
Back to business: The graph shows clear overlaps between PSP and the other groups, but the medians do differ to a statistically significant degree. The short arrow by the vertical axis points to a value of 85.06 ng/mg, which the researchers chose in retrospect as the best cutoff between normal and abnormal. Using that definition, the sensitivity of the measure was 100%, meaning that all eight participants with PSP had an abnormal result (that is, a value higher than 85.06). The same cutoff yielded a specificity of 70.6%, which is the fraction of the PD and HC participants with a normal result; in other words, the fraction that would be diagnosed correctly as “non-PSP.”
But if only 70.6% of the participants with “non-PSP” have a normal test result, that means that the other 29.4% have an “abnormal” result and would be falsely diagnosed with PSP. PD is about 20 times as common in neurological practice populations as PSP, so for every 1,000 patients who might have PSP or PD and see a neurologist, about 50 have PSP and 950 have PD. If you do the red cell test in all 1,000, that means that 29.4% of 950, or 279, will have an abnormal result. If all 50 with PSP also have an abnormal result, that totals 339 people with abnormal results, of whom 279 (a whopping 82%) don’t actually have PSP.
So, a neurologist seeing a result below that 85.06 cutoff would be able to reassure patient that they do not have PSP, with, of course, the usual precaution that outliers and lab errors do exist. A result above the 85.06 cutoff would prompt other diagnostic tests with greater specificity, although probably with greater expense, inconvenience and/or discomfort. I hasten to add that like any new research finding, this needs confirmation by other researchers using other, larger patient populations in all stages of illness.
You may recognize this result as the definition of a “screening test.” That’s a relatively inexpensive, convenient, safe and sensitive test suitable for use in large populations of asymptomatic or at-risk people. If a screening test is positive, further testing, or at least close observation, is advised. A good example is a routine mammogram, where a negative reading is great news and a positive reading prompts further testing. In this example, that testing usually results in a diagnosis of a benign cyst or scar or something else other than breast cancer, and the few women whose mammogram abnormalities turn out to be breast cancer and whose lives are saved by the ensuing treatment will be very glad to have had that screening test. A similar situation could develop for PSP once we have an effective way to slow or halt progression of the disease. That’s what the PSP neuroprotection trials currently under way hope to accomplish.
It seems unlikely from the new data that red cell alpha-synuclein concentration would ever offer enough specificity to diagnose PSP to the exclusion of non-PSP. But people with a positive test could then have, perhaps, an MRI, where certain arcane measures of the midbrain and basal ganglia could provide diagnostic information with the specificity for PSP that’s missing from the red cell alpha-synuclein test. In this way, the red cell alpha-synuclein is similar to neurofilament light, a protein elevated in the blood and spinal fluid in PSP but also in several other neurodegenerative diseases.
The senior author of the new paper is Dr. Andrea Quattrone, whom I know well and can vouch for. He is an internationally recognized leader in discovering diagnostic markers for PSP. The first-named author is Dr. Costanza Maria Cristiani.
More technical stuff in italics: Why should alpha-synuclein occur in elevated amounts in a tau-based disorder like PSP? Cristiani et al hypothesize that the red cells absorb most of their alpha-synuclein from the plasma (the liquid component of the blood) rather than being “born” with it in the bone marrow. They cite previous findings that excessive tau protein impairs the blood brain barrier, which could allow alpha-synuclein, an abundant protein in the brain, to leak into the blood, where it’s “scavenged” by the red cells. An obvious next step is to check other tauopathies such as Alzheimer’s disease for elevated red cell alpha-synuclein.
And now, on a personal note: My career as a researcher started in Parkinson’s disease and for a decade starting in 1986, I led the clinical component of the project that discovered that alpha-synuclein was related to PD. It began when I found and painstakingly worked up a large family with a rare, strongly inherited form of PD . That work, which included many collaborators I recruited in multiple institutions and countries, showed that family’s illness to be caused by a mutation in the gene encoding alpha-synuclein, which had not previously been suspected of any relationship to PD. Soon thereafter, others found alpha-synuclein as the major constituent of Lewy bodies (the protein aggregates of PD) in individuals with ordinary, non-familial PD without the mutation. Now, alpha-synuclein treatments and diagnostic tests are being developed for PD. So, if a critical diagnostic test for PSP, the disease to which I’ve devoted most of the more recent decades, should turn out to be based on alpha-synuclein, that would nicely satisfy the scientific narcissist in me.
Decades ago, the discovery that specific proteins aggregated in the brain cells of specific neurodegenerative diseases was a major advance. But like so many other scientific breakthroughs, it created another question: Why are there so many different clinical pictures among different people with the same neurodegenerative disease (like PSP) despite the fact that they all host the same aggregating protein (in this case, tau)? The ability of abnormal tau to “seed” the disease process into previously healthy brain areas is at the root of the disease process, but we’ve had scant clue as to how that works, exactly.
For PSP, the most important clinical variable is the eight subtypes (PSP-Richardson’s syndrome vs PSP-Parkinsonism vs PSP-progressive gait freezing, etc), and slightly less variable features are the onset age and rate of progression. In the past year or two, it’s become clear that the different subtypes tend to emphasize different areas of the brain, but that doesn’t explain why two people with the same subtype can have different onset ages and rates of progression.
This mystery became even more mysterious recently when a new electron microscopy technique called “cryo-EM” proved able to visualize individual protein molecules. It showed that for everyone with a given disease, the protein for that disease had the same misfolded shape. In other words, the tau molecule assumes the same rigid squiggle in everyone with PSP, a different rigid squiggle in everyone with Alzheimer’s, yet another in everyone with corticobasal degeneration, and so on. But that raised the question as to the reason for the variability among patients of the PSP onset age and rate of progression.
Now, researchers at the University of Toronto’s Rossy Centre, an institution dedicated solely to PSP research at the , have found new evidence supporting the old idea that the key may be in the “oligomers” or “high-molecular weight tau” or “HMW tau.” These are stacks of tau protein molecules small enough to remain dissolved in the brain’s fluids, as opposed to single molecules or the large, insoluble neurofibrillary tangles visible through a conventional microscope.
The top-line result was that the patients with more rapidly-progressive PSP and brain regions with the worst damage had higher levels of HMW tau. In a tour-de-force of lab experiments, the Toronto researchers also showed that:
HMW tau was more resistant to the brain’s mechanism for breaking down such protein clusters.
The study’s 25 PSP patients could be divided into high-, medium- and low-seeders based on the speed with which their tau converted healthy tau to their own misfolded form.
Tau with phosphate groups attached to amino acids 202 and 205 were least likely to form the HMW tau clusters.
The pattern of production of proteins (i.e., the “proteomics”) in the brain areas rich in HMW tau showed disruption of the brain’s adaptive immune system and two other cellular systems previously known to be related to neurodegeneration.
The importance of all this is that we now have a more specific idea of the structure of the most toxic form of tau aggregates and that boosting the brain’s adaptive immune system with medication could discourage the seeding of misfolded tau into healthy cells.
The study’s first author, Dr. Ivan Martinez-Valbuena, published an editorial in the journal Brain Pathology explaining all this in language that non-specialist scientists can understand.
The research paper itself is posted by the authors in bioRxiv (“bio-archive”) an on-line, open-access website for articles awaiting word from the peer-review process at a conventional journal. Its senior author is Dr. Gabor Kovacs, one of the world’s leading neuropathologists in the field of neurodegenerative diseases.
The last few posts have been about things at the macro level, from clinical trials to government action. Now, let’s dive back into some molecular biology — if you’re nerd enough for it.
Yesterday, a paper appeared from researchers at the University of Alberta, in Canada, led by Drs. Kerry T. Sun and Sue-Ann Mok, comparing the folding structure of normal and abnormal versions of the tau protein.
First, some background. You all know that proteins are strings of amino acids. The healthy adult human brain has six forms of the tau protein ranging in size from 352 to 441 amino acids. Tau’s normal job is to maintain brain cells’ internal structure and some other housekeeping tasks. Tau unattached to something else normally flops around in the cell’s fluid like a piece of overcooked spaghetti in boiling water. In PSP and the other tau-related disorders, tau becomes abnormally folded onto itself and forms toxic clusters that eventually clump further into neurofibrillary tangles. Those are visible through a microscope and are critical in the diagnosis of the “tauopathies” although the details of how misfolded or aggregated tau actually causes loss of brain cells remain unknown.
Some more background: Although over 99% of people with PSP have no mutations in the tau gene, there are 50 different mutations in tau that do cause neurodegenerative diseases, many of which closely resemble PSP. The most widely used experimental animal model for PSP has received a copy of a human tau gene with one of these 50 mutations.
The new project analyzed the folding structure of normal tau protein and samples of abnormal tau protein, each with one of the 37 most important tauopathy-causing mutations. It found that, at least as far as this lab technique could determine, no structural difference between normal tau and two of the most popular abnormal versions of tau used in research, the P301S mutation (where the amino acid proline at position 301 is replaced by the amino acid serine) and the R406W (arginine to tryptophan). Another mutation commonly used in animal models, P301L (proline to leucine) does alter the structure. That’s the form of tau addressed by the two monoclonal antibodies that AbbVie and Biogen, respectively, recently found did not help PSP.
Of the other 34 mutations tested, 12 produced no structural change and the location of the mutation had no discernible effect on the folding structure. Nor did the rate of aggregation influence the resulting structure.
Interestingly, one of those 12 producing detectable misfolding is the A152T (alanine to threonine) mutation, which is the only single-amino-acid substitution tau mutation we know of that increases the risk of “sporadic” (i.e., non-familial) PSP.
There are some caveats:
This study does not examine the effects of post-translational modifications (PTMs) on the folding structure of tau. Nor did it study the effects of the various mutations on the ability to accept PTMs. PTM’s are small molecules such as phosphate, acetate, methyl groups, sugars, and ubiquitin that can be attached to the protein in health to regulate its function, or as an effect of disease processes like PSP.
The study restricted itself to only one of the six adult human tau isoforms, called 0N4R.
The 0N4R form of tau has 383 amino acids (the others range from 352 to 441) and locations that can alter the folding pattern occur in only about 45 of those. So, as you’d guess, an amino acid substitution can change the chemical properties of a protein without changing its folding pattern. Another major issue is that many of those 45 misfolding spots are hidden inside the folded structure, obscuring them from the researchers’ analysis.
Despite these limitations, we can conclude that the various amino acid substitutions affect the misfolding pattern of tau in different ways. Any explanation of the cause of ordinary, sporadic PSP at its most profound molecular level can be guided by studying all of those misfolding patterns for hereditary PSP but will also have to take account of whatever bad thing the A152T mutation is doing – and that thing, according to this paper, is NOT to directly cause tau to misfold.
I watched a scientific presentation today in which the speaker started off by summarizing the leading theories of PSP’s pathogenesis. That means not the external influences such as the genes received from one’s parents or whatever toxins or other stresses might help cause PSP in susceptible people. Rather, it means the abnormal processes set in motion and operating inside in the brain cells leading to their dysfunction and eventually, their death.
Here’s a quick rundown for you:
Tau splicing. The tau protein is encoded by the MAPT gene, which has 14 sections called exons encoding separate fragments of the final protein. These protein fragments are then stitched together, but sometimes one or more of them is omitted by design. In healthy people, the product of exon 10 is included in about half of the final tau molecules, but in the tau tangles of PSP, that fragment is nearly always included. This makes the tau more likely to aggregate.
Tau post-translational modifications. Many or most proteins have very small molecules attached to them at specific points to regulate their function and direct their folding pattern. The abnormal tau of PSP has phosphate and other molecules in inappropriate places. This could help explain the abnormal folding, which in turn produces toxic aggregates.
Tau degradation. The normal “garbage disposal” systems of brain cells gets rid of proteins or organelles (the tiny structures in cells that perform specific functions) that are either overproduced, defective or just worn out. There are two basic kinds of such systems, the ubiquitin-proteasome system and the autophagy-lysosomal system. Neither works as well as it should in PSP. This allows abnormal tau and other toxic molecules to accumulate.
Intracellular tau spread. In many neurodegenerative diseases, the abnormally folded tau can travel from one brain cell to another, causing normal copies of those molecules to misfold in a similar fashion. This creates a kind of chain reaction spreading the damage widely. The misfolding pattern of the tau is specific to each of the tauopathies.
Mitochondrial dysfunction. The mitochondria are the organelles in the cells that harvest energy from sugars with the help of oxygen. In PSP, they function abnormally, possibly because of their own genetic mutations, possibly because their biochemistry is particularly sensitive to certain toxins in our environment. Mitochondrial dysfunction doesn’t just deprive the cell of energy – it also produces toxic compounds such as free radicals that damage other cell components.
Gene expression errors. The most recently discovered pathomechanism has to do with abnormal regulation of access of the cell’s protein-making machinery to the DNA “blueprint.” That process is normally regulated by proteins collectively called “chromatin,” which coat and intertwine with the DNA in the nucleus. One way the abnormality might work is that abnormal chromatin permits inappropriate access to certain genes that stimulate the immune system, producing a harmful inflammatory reaction in the brain.
All of these pathogenetic mechanisms except the first are currently being addressed by drugs in advanced stages of the development pipeline. I really don’t know which horse to put my money on.