Back in 2023, I posted an explanation of the ten PSP subtypes. The archetypal subtype, PSP-Richardson syndrome accounts for about half of all PSP and, in contrast to most of the other subtypes, has a rapid progression rate, a validated rating scale, and highly accurate diagnostic criteria. All of these features have led clinical trial sponsors to maximize their trials’ sensitivity and minimize their costs by restricting admission to people with PSP-Richardson. But developing better outcome measures for non-Richardson forms of PSP could change that practice.
A big step toward realizing this goal was published last week in the journal Neurology by a group at the Mayo Clinic in Rochester, MN. Led by first author Dr. Mahesh Kumar and senior authors Drs. Jennifer Whitwell and Keith Josephs, the study found that a good outcome measure for clinical neuroprotection trials in all PSP subtypes was to combine a measure of atrophy by MRI with a measure of clinical disability. This is a major advance.
The researchers performed brain MRIs at the start and end of a one-year period in 88 people with PSP and 32 age-matched controls. Of those with PSP, 50 had PSP-Richardson, 18 had “PSP-cortical” (three of the other nine subtypes) and 20 had “PSP-subcortical” (the other 6 of the subtypes). They had to lump the non-Richardson subjects using their subtypes’ general anatomical predilections because most of the subtypes were too rare to analyze on their own.
Calculating how much each of ten important PSP-involved brain regions had atrophied over the one-year interval allowed the researchers to identify which region(s) might best serve as markers of progression for each of the three groups when coupled with standard clinical measures. Those measures include such familiar instruments as the PSP Rating Scale and the Unified Parkinson’s Disability Rating Scale’s motor section as well as less familiar scales specific for cognition, gait, eye movement and speech. All the scales were administered concurrently with each of the two MRIs.
They expressed the sensitivity to one-year progression not by some abstract statistic, but by the number of patients needed in a double-blind trial to demonstrate with at least 80% certainty that patients on active drug enjoyed a 20% slowing of progression relative to the placebo group. (These specifications are typical for PSP clinical trials.) The better the measure’s performance, the fewer patients are needed.
And the award for Best Performance by an Outcome Measure in a PSP Neuroprotection Trial goes to . . . a combination of the rate of atrophy of whatever brain region shrinks fastest in the patient’s specific subtype and the PSP Rating Scale score.
The real significance of this study’s result is that using an outcome measure customized to each participant’s PSP subtype could allow trials to enroll not just people with PSP-Richardson, but also those with any of the other subtypes. That’s because the trial’s measure of success could be to compare each patient’s rate of progression during the trial to that of patients in the placebo group with the same PSP subtype.
This could double the number of people eligible to enroll in PSP trials, which means cutting the enrollment period in half, with commensurate reduction in costs for the sponsor. The hybrid measure is more sensitive to progression than the PSP Rating Scale alone, thereby reducing the number of patients required even more.
Both factors could lower the financial barrier confronting a company hoping to mount a trial for a promising PSP drug. That may be the most important bottleneck right now in the development of a treatment to prevent or slow the progression of PSP.s
That’s why this news is huge for PSP in general and for the “orphans” in particular.
“Pheno-“ is the Greek root for “outward appearance” and so far, PSP has ten of them. The differences arise from varying emphasis of the degenerative process among different parts of brain. While standard laboratory methods show that at the cellular level the pathology among the ten phenotypes is identical, a few details are starting to emerge using more recent and sophisticated techniques at the molecular level.
The most common PSP phenotype, called PSP-Richardson’s syndrome (PSP-RS), is the one Steele, Richardson and Olszewski originally described in 1964. The others were published piecemeal starting in the early 2000s. J.C. Richardson was the leader of the trio at the University of Toronto, a senior clinical neurologist who noticed an unusual form of Parkinsonism among his patients in the 1950s and 60s. John C. Steele was his trainee and Jerzy Olszewski was the neuropathologist who described the corresponding microscopical abnormalities. So, it’s altogether fitting and proper that Dr. Richardson should be honored in this way.
Prevalence of the phenotypes. The percentage of the PSP population with each phenotype has not been studied in a true community-based population. The published percentages vary widely across centers and are all from referral-based populations at research institutions, where unusual forms of diseases are over-represented to varying degrees. Even without that issue, several things make it hard to be sure of the prevalence of the various phenotypes:
It’s difficult to estimate the population prevalence of atypical cases of PSP from autopsy series because atypical cases are more likely to come to autopsy, and without autopsy, it’s hard to know that someone with atypical PSP really had PSP.
In their later years, all of the phenotypes tend to merge into a PSP-RS appearance, so the relative frequencies of the phenotypes may depend on the patients’ disease stage when the researchers evaluated them.
Clear diagnostic criteria do not yet exist for many of the phenotypes and many patients satisfy criteria for more than one, even in early stages. A method has been published for how to deal with this, but most of the publications antedate or ignore it.
Although the original differentiation of PSP-RS from PSP-P in 2005 did use a rigorous statistical technique called “factor analysis” to confirm that the two are distinct, this is not usually the case for the other phenotypes relative to PSP-RS or to one another.
Nevertheless, here are my very rough estimates of their contributions to PSP in general, based on a Gestalt impression of the literature:
Richardson’s syndrome
45%
PSP-RS
Parkinsonism
25%
PSP-P
Frontal
10%
PSP-F
Progressive gait freezing
5%
PSP-PGF
Speech/language
5%
PSP-SL
Corticobasal syndrome
3%
PSP-CBS
Postural instability
3%
PSP-PI
Ocular motor
3%
PSP-OM
Cerebellar
<1%
PSP-C
Primary lateral sclerosis
<1%
PSP-PLS
Many recent research articles group these into three categories based on their anatomical predilections in the brain: cortical vs subcortical. PSP-RS falls into neither of these because it has approximately equal degrees of both cortical and subcortical features. One important, practical reason for the grouping of phenotypes is to have groups large enough for meaningful statistical analysis.
PSP-Parkinsonism. The most common “atypical” (i.e., non-PSP-RS) phenotype of PSP is PSP-Parkinsonism (PSP-P). Relative to PSP-RS, it features more asymmetry, generalized bradykinesia, tremor, and levodopa responsiveness, and only later displays falls and cognitive loss. It is usually initially misdiagnosed as Parkinson’s disease. It has perhaps the slowest course among the PSP phenotypes, averaging about 9 years’ survival from symptom onset. This compares with about 6 years for PSP-RS and intermediate figures for the other phenotypes. In fact, the PSP-subcortical group as a whole has a similarly longer average survival duration than the PSP-cortical group as a whole or the PSP-RS + PSP-cortical groups.
PSP-progressive gait freezing. After PSP-P, the most common atypical phenotype is PSP-progressive gait freezing (PSP-PGF). In fact, most patients exhibiting only progressive gait freezing will eventually develop diagnostic features of PSP. The central feature of PSP-PGF is loss of ability to continue ongoing gait, especially after a pause, during a turn, or at a doorway threshold. In advanced cases, the patient cannot initiate gait at all. The picture also includes rapid, small handwriting and rapid, soft speech as frequent or severe features. The anatomic location of the pathology in such cases differs from that of PSP-RS in showing less involvement of the base of the pons (part of the brainstem) and of the dentate nuclei (part of the cerebellum).
PSP-speech/language. This is a composite category. In PSP-nonfluent/agrammatic variant of primary progressive aphasia (PSP-nfaPPA) speech is halting, with poor grammar, syntax, and pronunciation, but with normal comprehension and naming. A mirror-image variant called semantic-variant primary progressive aphasia (svPPA) features difficulty in naming with reduced vocabulary but with normal grammar and syntax. Together, PSP-svPPA and PSP-nfaPPA are referred to as PSP-speech/language disorder (PSP-SL).
PSP-corticobasal syndrome. CBS as a clinical syndrome (meaning a group of signs and symptoms that occur together, although the underlying disease may differ across patients) comprises highly asymmetric rigidity, slowed movement, and apraxia (loss of skilled movement), often with equally asymmetric dystonia (fixed postures), pyramidal findings (weakness and abnormal reflexes), myoclonus (small, rapid, irregular movements), and cortical sensory signs such as astereognosis (inability to identify objects by feeling them) and agraphesthesia (inability to identify figures traced on the skin). Aphasia (difficulty processing language) and other abnormalities localized to specific brain areas may also occur. Dysarthria (difficulty with pronunciation) can be prominent but gaze palsy, postural instability, and cognitive loss tend to be later and milder than in PSP-RS.
PSP-frontal. More formally called PSP-behavioral variant frontotemporal dementia (PSP-bvFTD or simply PSP-frontal), this phenotype features disinhibition, irritability, apathy, and loss of empathy for others, along with impairment in frontal “executive” functions such as ability to maintain attention, to follow instructions, to shift tasks on command, and to inhibit an ongoing action when appropriate. This is the core of the cognitive and behavioral deficits in PSP-RS, but when it appears first and remains worst, the term PSP-F is appropriate.
PSP-ocular motor and PSP-postural instability. Perhaps unsurprisingly given the cardinal features of PSP-RS, PSP can also take the form of a relatively pure ocular motor picture or a relatively pure picture of severe postural instability with falls and little else to suggest PSP. However, reported cases are very sparse to date. These have been designated PSP-ocular motor (PSP-OM) and PSP-postural instability (PSP-PI).
PSP-primary lateral sclerosis. The pathology of PSP can also produce the clinical picture of primary lateral sclerosis. PLS is one of the phenotypes of amyotrophic lateral sclerosis (ALS; Lou Gehrig disease) and can be difficult to distinguish from it, especially as ALS can produce frontal cognitive difficulties in many cases. The clinical picture of PSP-PLS is highly asymmetric and resembles that of CBS but with little or no cortical sensory loss (spatial sensation ability), dystonia (fixed postures), or myoclonus (very quick, small, irregular involuntary movements).
PSP-cerebellar. The classic lurching gait of PSP-RS has a cerebellar appearance, the speech of PSP has an ataxic (or drunken-sounding) component in many cases, and the ocular square-wave jerks of PSP occur commonly in cerebellar disease. For decades, these were considered minor and inconsistent features, but in 2009, neurologists in Japan described a PSP phenotype of PSP with involvement of the cerebellum at autopsy and early, prominent ataxia of the trunk and limbs. Although that original report found PSP-C in 14% of all people PSP at a Japanese center, the figure is much lower in Western populations for unclear reasons.
A word about drug trial eligibility. Until the cause of PSP in general is better understood, neuroprotection trials — those aimed at the fundamental brain cell loss rather than merely at ameliorating the symptoms — will continue to recruit only patients with PSP-RS. Why?
We don’t yet know if the non-PSP-RS phenotypes share – either with PSP-PS or with one another — the same molecular abnormality being targeted by the drug.
While the classic PSP pathology underlies close to 100% of PSP-RS, that figure is much lower for some of the other nine phenotypes. That means that non-PSP-RS may well be a non-PSP pathology, and that admitting participants with non-PSP-RS to a drug study runs a risk that some may not have PSP at all. This could obscure any benefit the drug may have unless the trial is prohibitively large.
The main outcome measure for nearly all PSP trials is the PSP Rating Scale, which was designed for, has been validated for, PSP-Richardson’s syndrome alone.
Trials to slow the progression of PSP use the rate of progression as the “outcome variable” of the trial. As noted above, the phenotypes do vary in their expected survival durations, and by inference, their progression rates. Therefore, including phenotypes with different inherent rates of progression would require a larger, longer and more expensive trial. One solution would be to make sure the active drug group and the placebo group receive similar proportions of the various phenotypes (a time-proven technique in trial design called “pseudo-randomization”). But this doesn’t solve the four preceding problems.
As PSP-RS progresses faster than the other nine phenotypes, a trial enrolling only that phenotype can reach a result in a shorter time or with fewer participants. This isn’t only financially advantageous for the trial’s sponsor. If the drug is ineffective or harmful, fewer patients will have been exposed to it, and if it’s effective and safe, it will reach market approval more quickly.
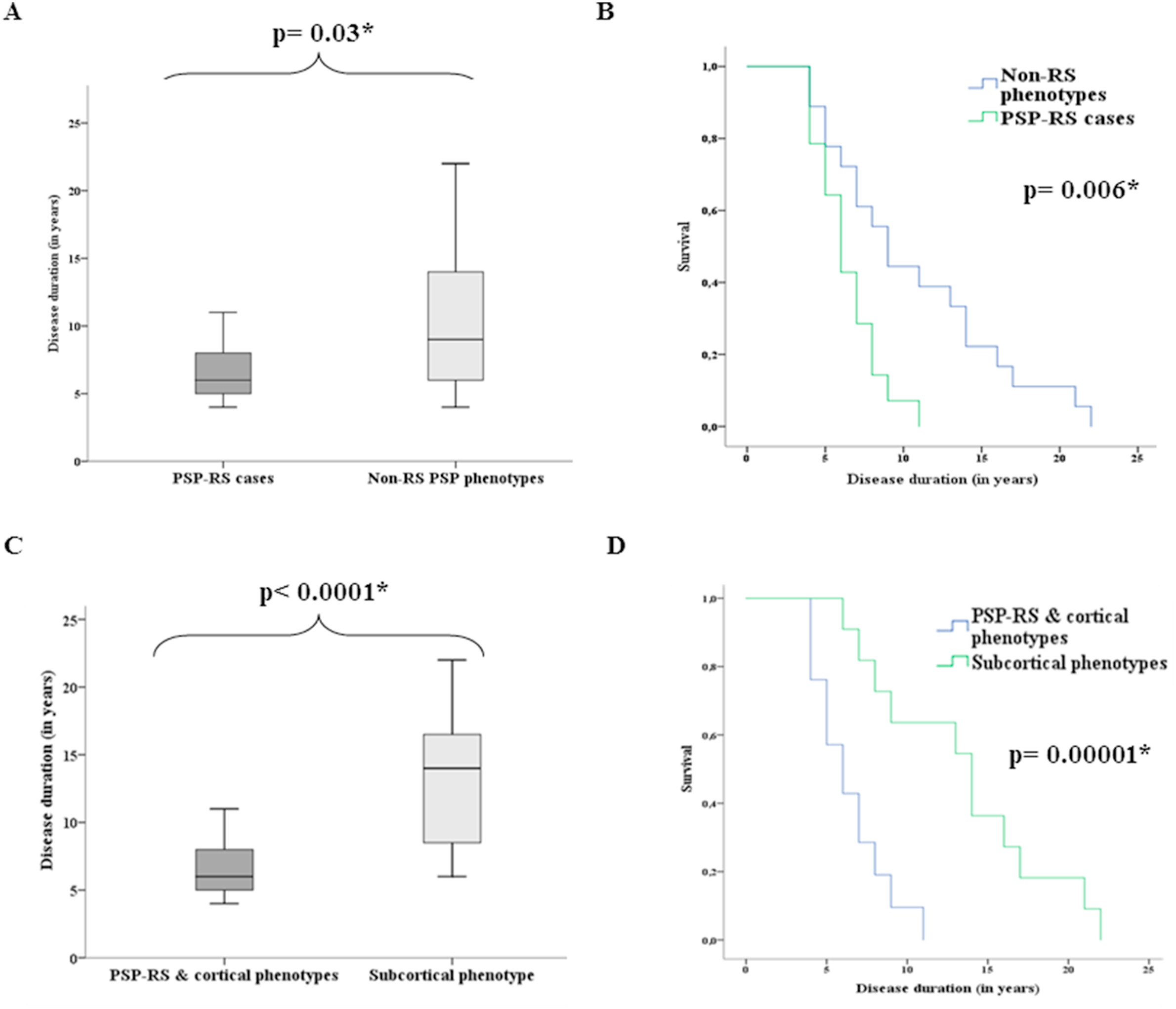
These graphs are from a review of records of 32 people with autopsy-proven PSP in Spain and Germany. The lead author was Dr. Mar Guasp of Hospital Clínic de Barcelona and the senior author was Dr. Yaroslau Compta of the same department. They show the difference in survival from initial symptom to death between the PSP-RS and non-PSP-RS (top) and between [PSP-RS + PSP] and PSP-cortical (bottom). Graphs A and C: For each box, the horizontal line is the median, the upper and lower borders are the 25th and 75th percentiles and the ends of the “whiskers” are the highest and lowest values. Graphs B and D show the same thing in the form of “survival curves” or “Kaplan-Meier curves.” The vertical axis shows the fraction of the original patients still living at the time (post-onset) shown on the horizontal axis. The p values are the likelihood that the difference could have happened by random chance. The asterisk indicates that this likelihood is low enough for the difference between the groups to be considered “statistically significant.” (Statistical veterans: Sorry to belabor this for the benefit of the statistical novices.)
To-do list:
Let’s figure out why the disease spreads through the brain in PSP-RS and the other PSP-cortical phenotypes more quickly than in PSP-subcortical. Efforts to do that have in fact begun and could provide the key to the whole puzzle of PSP.
Let’s agree on a way to enroll people with non-PSP-RS phenotypes into clinical trials. Current efforts to diagnose PSP using tau-based positron emission tomography (PET) and measures of tau in skin, blood or spinal fluid could potentially identify people with PSP other than PSP-RS who could potentially join a trial.
Let’s educate neurologists to identify, or at least to suspect, the non-PSP-RS phenotypes. This would allow them to avoid or delay fruitless diagnostic testing and to provide their patients with useful prognostic information.
When you design a research project in PSP, it’s important to make sure that everyone in the subject group with PSP in fact has PSP. Otherwise, you degrade the statistical power of the trial to detect any benefit of the treatment. The standard diagnostic criteria for PSP (called the “NINDS-SPSP Criteria” and spearheaded by Irene Litvan, MD, now of UCSD) were published in 1996 and have worked well for that purpose; Their positive predictive value (the fraction of patients satisfying the criteria who actually have PSP) and specificity (the fraction of those without PSP who fail to satisfy the criteria) are close to 100%.
But as we now enter the new era of trials of experimental neuroprotective treatment for PSP, we would like to diagnose the disease at an earlier stage, when such interventions are most likely to be effective, and the NINDS-SPSP Criteria don’t do that so well, with a sensitivity of about 80% overall, certainly lower in early cases. Another shortcoming is that the various phenotypes of PSP that have been described since 2005 won’t in many cases satisfy the criteria, which were designed for the “original flavor,” now called PSP-Richardson’s syndrome.
So time has marched on and we need a new set of criteria. Günter Höglinger, MD, Professor at the German Center for Neurodegenerative Disorders in Munich and probably the world’s leading clinical researcher in PSP, organized an international effort to revise the criteria. I’m privileged to serve on the four-person Steering Committee. A year ago we started to hash things out by email and conference calls, using the published articles on clinical features of PSP that use either autopsy or the NINDS-SPSP Criteria as a gold standard. The group, comprising 33 people from 11 countries, met in Munich on March 9 and 10 to turn our rough draft into a final version suitable for submission to a journal for peer review.
The new criteria recognize the various phenotypes of PSP. They are PSP-Richardson syndrome (about 55% of all PSP), PSP-parkinsonism (30%), PSP-frontal dementia (5%), PSP-ocular motor (1%), PSP- pure akinesia with gait freezing (1%), PSP-corticobasal syndrome (1%), PSP-progressive non-fluent aphasia (1%), and PSP-cerebellar (<1%). The remaining few percent are combinations of these or still-unrecognized forms.
The new criteria also delineate various “oligosymptomatic” or “prodromal” (the wording remains unsettled) forms, which may or may not develop into one of the diagnosable phenotypes. For example, there is now evidence that someone in the PSP age group with gradually progressive gait freezing for several years and a normal MRI, even without other abnormalities, will almost always prove to have PSP. The same is true for someone with bilateral rigidity and bradykinesia who fails to respond to levodopa and has some sort of nonspecific, undiagnosable visual symptoms or dizziness. Neither of these patients would satisfy the proposed new criteria for any of the PSP phenotypes, but they may still be worth identifying for inclusion in a longitudinal cohort study of people who are at risk of developing PSP. Our new criteria do that.
I’ll keep you updated.
As it turns out, PSP comes in many clinical flavors. Back in the 80s I remember some patients whose illness looked like Parkinson’s until I realized that they weren’t responding to my levodopa prescriptions, at which point I repeated a careful ocular motor exam and found square wave jerks and slow downward saccades. I also remember one member of my first series of 41 patients with PSP from 1988 with severe gait apraxia and freezing as his most disabling feature.
Then, in 2005, David Williams and colleagues, mentored by Andrew Lees at Queen Square, published what is probably the most important clinical paper on PSP in the half-century since Steele, Richardson and Olszewski. That work delineated and named PSP-Richardson syndrome (PSP-RS ) and PSP-parkinsonism (PSP-P). This wasn’t just a new way to slice a clinical spectrum sharing the same basic pathology; the two variants actually had statistical differences by cluster analysis. This suggests that they differ at the pathoanatomic level. They even differed in the ratio of 4R/3R tau. (It turns out that the predilection of PSP’s tangles for 4R tau is driven by RS.)
Since then, a cornucopia of low-frequency clinical variants meeting pathoanatomic criteria for PSP has been described. In approximate descending order of prevalence after RS and PSP-P are corticobasal syndrome, postural Instability, pure akinesia with gait freezing, frontotemporal dementia, ocular motor predominance, progressive non-fluent aphasia, semantic dementia, and a cerebellar variant.
The clinicopathologic studies are only starting to appear, but it’s likely that they will all turn out to emphasize different cells, nuclei and brain regions. We will also probably see some subtle molecular differences among them (presaged by the 4R/3R difference between RS and PSP-P).
That sounds like different diseases to me. Different diseases shouldn’t be combined in treatment trials, genetic analyses or descriptive studies. What a mess.
Or is it? Maybe we don’t need to find causes and cures for each PSP variant individually. As they’re all tau aggregation disorders, maybe they will all yield to the same prevention. Maybe the mechanism of prion-like spread, by now pretty much a textbook verity, will apply not only to all of the “pure tauopathies” (and it’s not yet clear that all of the PSP variants are in fact pure tauopathies) but to all of the protein-aggregation-based neurodegenerative disorders. If it does, then poisoning that process could be the grand unified answer to Alzheimer’s, Parkinson’s, ALS, and PSP in all its malign variety.