Since 2014, I’ve been trying to find the cause of a geographical cluster of PSP in northern France. It’s the only documented PSP cluster known. The problem was difficult enough, but now the cluster has mysteriously disappeared.
The clinical aspects of the cluster are detailed in this 2015 paper. Here’s the executive summary: In 2005, Dominique Caparros-Lefebvre, MD, a geriatric neurologist with experience in PSP research, arrived at her new practice position in Wattrelos, France, an industrial suburb of Lille. By 2007, she started to notice more PSP than expected and developed an excellent database. She diagnosed 100 patients over the next decade. In 2013, she invited me to help her find the cause, as I had had some experience in the epidemiology of PSP. I calculated the observed-to-expected incidence ratio of PSP to be 12.3 in Wattrelos and its neighbor to the south, Leers. Most clusters of chronic diseases such as cancer have ratios much lower, in the range of 1.5 or 2.0. So this was a major cluster.
PSP in the 100 patients has differed only slightly from “sporadic PSP,” with more PSP-Parkinsonism than PSP-Richardson syndrome, and an older mean onset age, 74.3. The 13 autopsied cases show typical PSP, with the expected 4R tau and the H1/H1 genetic haplotype. That work was done by a very accomplished research team at the University of Lille led by Luc Bueé, PhD and Vincent Deramecourt, MD, PhD. No other molecular genetic workup has been performed to date, but none of the affected persons were related to one another and among the patients are 7 Algerian immigrants, a strong point against a genetic origin.
Wattrelos and Leers have extensive chemical contamination, especially by metals from an ore extraction plant that operated in southern Wattrelos for most of the 20th century. Huge piles of spent chromate and phosphate ore, now covered, remain on the plant’s property, which has been converted into to a public park after mitigation efforts between 2000 and 2010. Only 2 of the 100 patients worked in the chromate/phosphate ore plant, but soil from the area adjacent to the slag heaps has been used as fill in construction and road maintenance over a wide area. Furthermore, multiple chemical-related industries such as tanning and dyeing formed the base of the town’s economy for many decades.
So, the obvious culprit has been metals. Chromium is a carcinogen but not a good candidate as a direct neurotoxin, as its most common form, hexavalent chromium, does not cross the blood brain barrier. Nor is phosphorus a good candidate, but phosphate ore often contains important levels of other metals.
In France, growing one’s own herbs and vegetables is a common practice, even in densely urban areas. Dr. Caparros suspected thyme, a widely used herb in French cooking that avidly absorbs metals from the soil. The French government’s soil data and our analysis (by my Rutgers colleague Brian Buckley, PhD) of home-grown thyme samples from Wattrelos suggested that arsenic, cadmium and nickel were the most likely possibilities.
In 2016, I recruited a team of neuroscientists led by Aimee Kao, PhD of UCSF, with skills in stem cell models of PSP and access to stem cells with PSP-related tau gene mutations. As an initial project, they created brain cells with a rare PSP risk mutation (to create a “background” vulnerability) and exposed them to chromium, cadmium and nickel. They did the same experiment with cells from the same PSP patient except that the PSP risk mutation was converted to normal using CRISPR. They found that some of the same damage seen in PSP — aggregation of tau and evidence of apoptotic (i.e., programmed cell death) in the exposed cells with the mutation. But those abnormalities are not specific for PSP. We published that in 2020 and unfortunately, I couldn’t keep that team together for follow-on projects. Equally unfortunately, the local French human research authority would not allow Dr. Caparros to perform further field work that might have pointed to a specific metal and route of exposure.
So why aren’t clusters of PSP seen in the many other places in the world where those metals contaminate the environment? My own pet theory was that toxicity from multiple metals acting in concert is needed, and no place other than Wattrelos/Leers has a combination of so many metals in one spot together with a physician able to diagnose PSP as well as Dr. Caparros. So, one of the follow-on projects might be to repeat the lab experiments with combinations of the same and other metals that are known to occur in the environment, either in Wattrelos/Leers or elsewhere, either as a result of industrial pollution or naturally occurring.
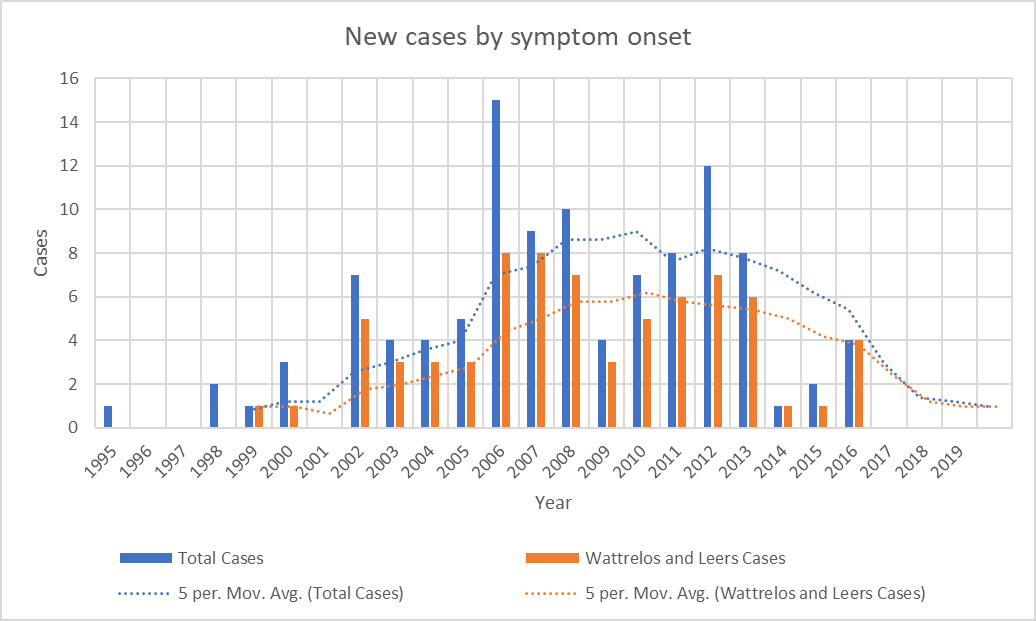
As I was starting to make plans for such a project with lab colleagues at Rutgers, it became clear that the number of patients whose onset occurred since 2013 has been declining. The most recent onset date of the 100 patients is 2016. This is not how a cluster of toxic (or genetic) cause should act. It’s possible that the mitigation efforts on the two slag heaps reduced the rate of entry of chromium and phosphate into the local environment, but the metals were pervasive in the area and presumably remain so.
—-
—-
The unexplained abatement of a geographical or temporal disease cluster speaks for an infectious cause. The salient example of an infection causing a temporal cluster of a neurodegenerative tauopathy is postencephalitic parkinsonism (PEP). That cluster started 2 years before and ended a decade after the great “Spanish” flu pandemic of 1918-1920 and is independent from it. PEP was a chronic, levodopa-responsive parkinsonism that affected people of any age who recovered from an encephalitis that was presumably viral, but the specific virus has not been identified. At autopsy, the brain showed neurofibrillary tangles not very different from those of PSP. The last patients with PEP died before modern molecular techniques were available, so its cause may never be known.
Could the cause of the Wattrelos/Leers cluster have been a virus? True, there seemed to have been no antecedent encephalitis, but it may have been mild, self-diagnosed as a cold or the flu, and forgotten by the time Dr. Caparros saw the patient decades later. But there need not have been any clear acute-phase symptoms at all. The virus could have set up a slow process of damage involving tau aggregation, starting with inserting its own genetic material into that of the host. Or the initial infection could have altered the patient’s immune system in a way that encouraged (or allowed) the pathology of PSP to develop. Let’s not forget that disordered immune modulation is one of the up-and-coming theories of PSP-causative factors.
If a virus contributed importantly to the cluster, could ordinary, “sporadic” PSP outside of the cluster be the result of a similar virus? Or maybe sporadic PSP is caused by the same virus without the predisposing local factor of the unusual metals exposure. Or maybe a virus infected the gut microbiome of the Wattrelos population in a way that increased PSP risk. I could go on.
We know that at least one neurodegenerative condition, sporadic Creutzfeldt-Jakob disease, is caused by an infectious agent (in this case the prion protein) without geographical or temporal clustering. The idea of a virus or prion as a cause of PSP is not new, and previous attempts to prove that hypothesis starting in the 1970s have been negative. But the technology for finding viral fingerprints has improved markedly since then.
I’ll try to get some of the research honchos I know interested in this theory and get back to you.