You might be interested in a review article that came out yesterday in a journal called Alzheimer’s & Dementia: Translational Research & Clinical Interventions. It discusses the unique challenges in the design of clinical neuroprotection trials in PSP.
(“Neuroprotective” treatments, as many of these posts have explained, attempt to slow the rate of worsening after the diagnosis is made. Measures to prevent disease in healthy people, like a flu shot, is called “preventative” and those to repair damage are “restorative.” PSP is further from either of those than it is from protective treatment.)
The article points out that a major goal for PSP trials is reducing the number of subjects needed: the “N.” That’s because the rarity of PSP makes it difficult to find qualified participants, and fewer of those means longer recruitment time and sometimes, longer trial periods. One way around that problem is more sensitive outcome measures, and that’s one goal of the search for better biomarkers.
Another way around the problem of keeping the size of PSP trials manageable is to form an expectation at the study’s outset, based on existing research, of each subject’s likely progression over the next 12 months, calculate what percentage of that expectation actually occurs, and then compare active drug with placebo groups on their respective averages of that percentage.
Another approach is to specify disability milestones such as progressing from independent gait to needing occasional assistance, or from a walker to a wheelchair, and then comparing the active drug and placebo groups according to how many milestones they reached over the 12 months. One type of milestone is called the “minimum clinically important difference.” That’s the main driver of the article and I’ll explain it in a future post.
The authors of the new paper are myself and a statistician, Dr. Ronald Thomas, at University of California, San Diego. It’s written at a level that most of you will be able to understand and I’d love to hear your honest reactions in the comments area.
An experimental, orally-administered drug called ARV-102 will enter clinical trials for PSP very soon.
Last month, a small, Connecticut-based biotech company called Arvinas announced favorable results of a Phase 1 safety/tolerability study in people with Parkinson’s disease and expects a Phase 1b trial in PSP to begin in the “second quarter” of 2006.
(For most drug companies, the quarters conform to the regular calendar, but for others, it’s their fiscal year, and I don’t know which applies to Arvinas. Phase 1b studies, like others in Phase 1, focus on safety rather than efficacy, but include patients with the target disease rather than healthy volunteers.)
ARV-102 is a member of a drug class called PROTACs, and here’s how they work, as described in my post from October 20, 2021 (yes, five years ago):
Another approach that sounds like science fiction is “proteolysis-targeting chimeras (PROTAC) molecules, first developed in 2001. These are hybrids of two small molecules, one of which can bind to tau (in this case) and the other that is recognized by the brain cells’ internal garbage disposal called the ubiquitin-proteasome system. Unlike antibodies, PROTACs can easily enter brain cells. Their only human application so far has been in oncology, but experiments in mouse tauopathy models are proving successful.
The company has made an animated video showing how ARV-102, which you can find here (click the “watch video” button). Arvinas has several other kinds of PROTAC under development, most of which are for forms of cancer.
As the video states, PROTAC drugs attach to the target protein like antibodies, but unlike antibodies, are not consumed by destroying a single molecule. Rather, they are like enzymes in that they facilitate chemical reactions hundreds of times until they wear out.
My explanation in italics above mentions tau as a potential PROTAC target, but ARV-102 targets a different protein called leucine-rich repeat kinase-2, or LRRK2 (pronounced, “lark-two”). Like all other kinases, it attaches phosphate groups to proteins as a way to regulate their function. The proteins regulated by LRRK2 are involved in a few dozen important processes in brain cells, the most important of which for PSP is probably an enzyme called Rab GTP kinase. Rab is a “master regulator” of the cell’s ability to direct tiny packets of compounds, called vesicles, from where they’re made to where they need to be. That’s called “vesicular trafficking.”
Three of the leading reasons to suspect that LRRK2 is misbehaving in PSP are:
One of the gene variants most consistently associated with the risk of developing PSP is called STX6. After Rab (see above paragraph) guides vesicles to their destinations, they are fused in place by the protein encoded by STX6.
One variant of LRRK2 has been found to be strongly associated not with the risk of developing PSP, but with shorter survival of those with PSP.
Another LRRK2 variant called G2019S, which by itself causes a form of hereditary Parkinson’s disease, can in rare cases cause the signs and symptoms of PD during life but with the microscopical pathology of PSP.
The scientists among you are now thinking that this evidence for a LRRK2-PSP link is sort of indirect, as opposed to the direct evidence linking LRRK2 with Parkinson’s. But the people at Arvinas may feel that PSP is also worth a try because the need there is so great and that success in PSP could open up potential for other tauopathies like Alzheimer’s.
Let’s be glad that Arvinas has joined the list of companies with innovative drugs going to bat for PSP. (Disclosure: No, I don’t consult for them nor hold their stock nor any of that other stuff that could explain my optimism.)
One of this blog’s frequent commenters — OK, it’s my pal Jack Phillips, CurePSP’s Board Chair — prompted by my 3/8 26 post, has asked which of the three PSP Trial Platform drugs to bet on. That’s a tough one, partly because unlike racehorses, each has a different mechanism of action, so it’s an apples/oranges/peaches comparison. But as long as I don’t have to worry about peer review of my blog posts, here’s what I’m thinking at this point:
The AADvac anti-tau vaccine induces one’s immune system to make anti-tau antibodies, so one might expect it to do no better than the two failed passive (i.e., directly infused) monoclonal antibodies from a few years ago. But the antibodies formed in response to AADvac recognize the middle part of the tau protein, while the monoclonals recognized the initial (i.e., N-terminal) end. There’s good evidence now that the toxic part of abnormal tau is in AADvac’s middle-domain wheelhouse. So, the questions now are:
Has tau already done its dirty work before reaching the form or location susceptible to the antibody?
Most of the damage done by tau happens inside the brain cells, where antibodies can’t reach. The hope is to pick off the abnormal tau in transition from one brain cell to another. That works in mice with an abnormal version of the tau gene that causes a familial form of frontotemporal dementia with Parkinsonism. But FTDP isn’t quite PSP and mice aren’t quite people.
LM11A-31 is very different. It enhances the brain’s ability to repair existing damage. It has shown benefit in a number of different animals models of different diseases with different aggregating proteins (or with none). In humans with neurodegenerative diseases, the only published experience is in Alzheimer’s disease, where the benefit was modest, though the study was too small to assess efficacy in a valid way. My concerns are that:
The drug’s modulation of the cells’ compensatory mechanisms might be too subtle to stand up to the onslaught of misfolded tau and other perturbations present in PSP.
Starting from an early stage of involvement in PSP, brain cells transmit misfolded tau to other cells. It’s possible that this happens before the cells have lost much of their functional abilities, perhaps before the mechanisms that LM11A-31 modulates become relevant.
AZP-2006 improves lysosomal function, thereby helping the cells dispose of tau that’s overabundant, misfolded, aggregated or excessively phosphorylated. I personally favor that idea for three main reasons:
The high frequency of co-pathology (where the tauopathies have mild levels of other aggregating proteins) suggests that specific defects in a shared garbage disposal system affect specific combinations of proteins. This in turn implies that if the predominantly affected protein is tau, then a tauopathy develops, with a few aggregates of other proteins such as α-synuclein, TDP-43 and others. I’d done research on the PSP cluster in a group of towns in northern France with severe ground contamination by multiple industrial metals. Lab experiments (performed in collaboration with a team under Drs. Aimee Kao and Carolina Alquezar at UCSF) have suggested that some of the metals in that environment can damage the disposal mechanism without affecting the production of tau itself. That suggests that a treatment like AZP-2006 aimed at that mechanism could work.
AZP-2006 has already shown evidence of an ability to slow the progression of PSP. That trial, designed to detect safety and not efficacy, included only 24 patients on the active drug and lasted for only six months. So, the slowing of progression relative to placebo did not reach statistical significance. But its magnitude was most impressive. https://movementdisorders.onlinelibrary.wiley.com/doi/epdf/10.1002/mds.70049
So, my analysis gives AZP-2006 a slight edge among these three. But that’s based partly on results of my own research, so I have a sentimental bias. Then there are other drugs in the pipeline, like:
NIO-752 (a tau-directed anti-sense oligonucleotide to reduce tau production)
FNP-223 (an inhibitor of an enzyme that allows phosphate group to attach to tau)
GV-1001 (mostly an anti-inflammatory to quell one important step in the disease process)
Besides, much smarter people than I have been crashingly wrong in predicting clinical efficacy of drugs. But it’s a good mental exercise to think about it.
Welcome news on the PSP Platform Trial (PTP). As you recall, that’s an NIH-supported organization at about 50 US academic medical centers designed to test three anti-PSP drugs from three different drug companies simultaneously using one placebo group and one administrative apparatus. The most recent expectation is for a start in the third quarter of 2026.
The news is that the PTP has now added the last of the three initial drugs, provisionally called LM11A-31.
It’s an oral drug that interferes with the degenerative scripts in brain cells, potentially giving them a chance to repair themselves. It accomplishes that by activating a receptor protein on the surface of brain cells called p75NTR, which modulates the cell’s response to various kinds of insults.
As you’d guess, this mechanism could apply to many other brain diseases. In fact, LM11A-31 is being investigated in animal models for Alzheimer’s, Parkinson’s and Huntington’s diseases, HIV dementia, stroke, traumatic brain injury and fetal oxygen deprivation. However, the lab animal evidence for a strong anti-tauopathy effect seems particularly strong. I’ll let the mavens explain (nerd alert):
“In AD and tauopathy mouse models, oral administration of LM11A-31 reduced excess activation of enzymes contributing to tau post-translational modifications, accumulation of multiple forms of pathological tau species and tau seeding activity, reduced elevations in multiple microglia and astrocyte markers, and decreased the loss of dendritic spines and synapses while improving performance on hippocampal-dependent memory tasks,”
The paper from which I copied/pasted that was a 6-month, Phase 2 trial of LM11A-31 in 242 people with Alzheimer’s disease. A third of that group received placebo. The drug showed acceptable safety and tolerability, with the incidence of side effects of the 200 mg dose about the same as that of the placebo and the 400 mg dose moderately higher. A secondary aim of the trial was to look for indirect evidence for slowing of progression in various neurodegeneration-related imaging and spinal fluid measures. Such evidence, promisingly, did occur more frequently in the participants on the active drug than in those on placebo.
But the Phase 2 AD trial’s potentially great news is that the rate of worsening of the cognitive loss itself as measured by each of three standard tests did seem to slow down a bit. The magnitude of that benefit did not reach statistical significance in the 6 months available, but AD trials designed to detect efficacy (as opposed to safety) generally need a year or two to have much chance of showing a statistically significant effect.
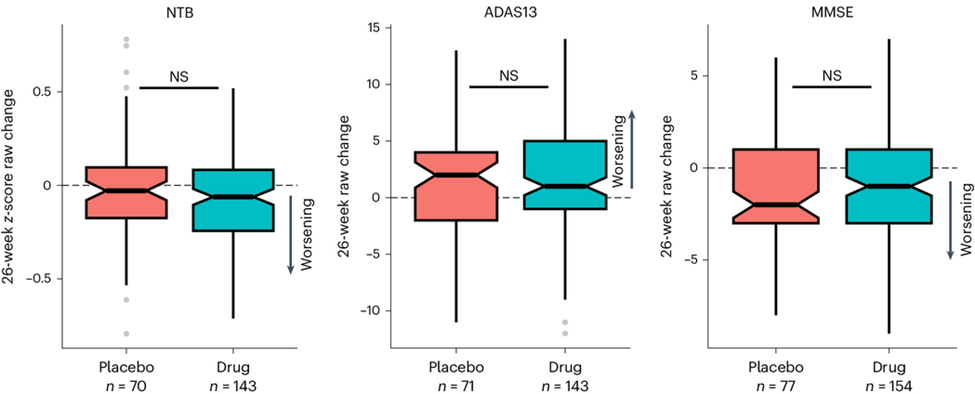
The graphs above show that trial participants with AD who received LM11A-31 progressed a bit more slowly on each of the three cognitive tests than those on placebo. However, none of the differences reached statistical significance.
NTB: Neuropsychological Test Battery; ADAS13: 13-item version of the Alzheimer’s Disease Assessment Scale; Mini-Mental Status Exam (from Shanks HRC, et al. Nature Medicine, 2024). https://www.nature.com/articles/s41591-024-02977-w
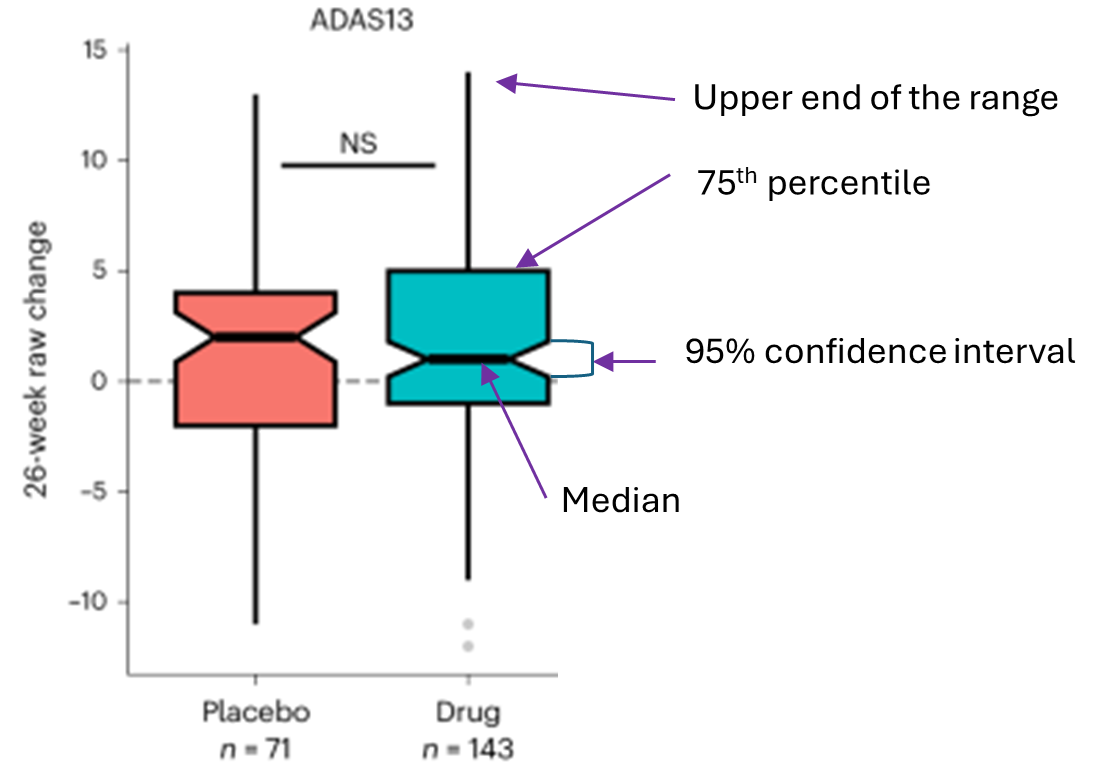
The diagram below explains the above graph’s components:
ADAS13 is the cognitive test.
The upward direction is worse. Downward is better.
NS means no statistically significant difference between the active drug and placebo groups.
The “raw” change (vertical axis label) means that the scores were not adjusted for potentially confounding factors. Such adjustment of efficacy results is routine in trials designed to assess efficacy, but not in this trial, which was designed to assess safety and tolerability.
The PSP Trial Platform’s study will also be a Phase 2 trial like the AD trial, but it will be scheduled for a full 12 months rather than six. If it starts in the third quarter of 2026 as planned, the study would be completed in mid-2029. At that point, the FDA could decide to approve the drug without a large Phase 3 trial if they feel that the results of the Phase 2 are sufficiently favorable and if there’s no other neuroprotective drug for PSP on the market at that point. The other two drugs in the PTP’s inaugural round will be AZP-2006, which enhances the breakdown of abnormal tau by the lysosomes, and AADVac1, an anti-tau active vaccine.
The sponsor of LM11A-31 is a tiny company called PharmatrophiX, Inc., founded by the scientist who discovered the drug, Frank M. Longo, MD PhD, of Stanford University. https://www.pharmatrophix.com/
Here’s great news: The new Federal spending bill, which the president has now signed into law, includes a continuation of Medicare’s payment for tele-health visits, including audio-only visits, through December 2027. It essentially cancels, for now, the planned return to Medicare’s pre-Covid telehealth policy.
This is especially important for those with PSP, where neurologists familiar with the condition are few and far between and where many of those affected have difficulty traveling, even by car.
PSP is in a better position now than it was pre-Covid to adapt to telehealth. Two versions of the PSP Rating Scale that omit the items difficult to perform by video were validated in 2023. Disclosure: I was a co-author. The original version of the PSPRS, published in 2007, has 28 items and I’ll refer to it as the “PSPRS-28.” (Disclosure: I developed it and validated it with the help of statistician Pam Ohman-Strickland). One video-friendly modification, the PSPRS-25, omits the three items that require the “laying on of hands,” to use a term that shows my age. The other, the PSPRS-21, omits those plus the four others relating to eye movements and dystonia. With the sub-optimal technical situation in the patient’s home, video’s image resolution is not adequate to assess those items, even with the caregiver’s assistance.
The procedure applied the PSPRS-25 and PSPRS-21 to records from PSPRS-28 administration to two patient groups. One was the placebo group from the 12-month davunetide PSP study published in 2014 . The other was my own patients with PSP seen from 1994 to 2020, where I applied the PSPRS-28 at each visit. The result was that there was excellent correlation with the PSPRS-28 in the 12-month trial database and with long-term prognosis in the long-term database. That project was led by Drs. Alexander Pantelyat of Johns Hopkins and Anne-Marie Wills of Mass General.
Other support for video visits for PSP was provided by a study from 2020 where two neurologists independently evaluated a series of patients with various atypical Parkinsonian disorders by live video and compared diagnoses. The degree of agreement was excellent, with a kappa score (the standard statistical test for inter-rater agreement) of 0.83. A kappa of 1.0 is perfect agreement and anything over 0.75 or 0.80 is considered excellent. That study is from the University of Rochester, led by Drs. Christopher Tarolli and Jamie Adams.
It’s great to know that an accurate PSP Rating Scale exam is feasible by video, and to know that neurologists almost agree on their video diagnosis of atypical Parkinsonian disorders. But there’s a lot more to the care of someone with PSP than diagnosis and symptom rating. But hey, maybe tele-health, with a hand from AI, will learn to do all that, too. Give it a few years.
Every so often I’ll make the rounds of what’s out there on the Web by way of PSP information. So I thought a nice use of a cold day would be to share my latest observations with you all.
Every proper scientific literature review starts with a description of methods. Mine was to type “progressive supranuclear palsy” into Google and to go with the first seven sites listed. They’re allegedly the most-visited unless the sponsor has paid to be listed on top, which is the case for one of these. I added CurePSP’s main page on PSP information, which didn’t make the top seven probably because people new to PSP have heard of the other organizations but not CurePSP. I also added what ChatGPT had to say, which was a high-level summary that offered users some search terms for further details.
Clearly, CurePSP and Wikipedia were the winners, but many users could be stressed or confused by their level of detail. If that’s you, my next recommendation would be ChatGPT, but if you don’t like their very terse, outline style, I’d go with Penn Medicine. But I advise checking out more than one.
Again, like every proper scientific paper, this one has a disclaimer statement, which is that I helped write the material in the CurePSP website and am the guy lecturing in its videos mentioned here.
The sources are listed alphabetically.
Source(alpha-betical)
Word count
Number of links to more info
Accuracy(absence of incorrect state-ments)
Compre-hensiveness (relative to others here)
Technical level (relative to others listed here)
Quality/quantity of illustrations, charts, graphs, videos
Months since last update
Com-ments
ChatGPT
302
0
9
6
Moderate
None
0
Offers examples of search terms for more detail
CurePSP
4,116
Dozens
10
10
Moderate
2 videos of lectures, 1 of an affected family
Continual
One page in a very large website on PSP, CBS & MSA
Davis-Phinney Founda-tion
1,461
24
6
5
Moderate
none
29
Paid for Google search place-ment
Mayo Clinic
1,477
3
9
7
Easy
none useful
24
No statistics; 8th-grade reading level
NIH
1,803
10
9
5
Moderate
none
10
No statistics or lab science
National Health Service (UK)
708
8
9
5
Easy
none
6
Very super-ficial
Penn Medicine
798
9
10
4
Easy
none
?
Basic but well-pre-sented
UC San Diego
955
0
9
8
Difficult
Includes excellent video interview with expert
?
Outdated; few topics in text but more in video
Wikipedia
4,593
Dozens
10
10
Difficult
A useful table
Continual
Missing non-tau genetics & current diagnostic criteria
A reader just commented on the need to improve public awareness of PSP and asked how that could be accomplished.
CurePSP devotes a large chunk of its budget and staff to raising awareness of PSP. For example, a few months ago, some of CurePSP’s leaders, including myself, met on Capitol Hill with 12 members of the House and Senate from both parties to make them aware of PSP, CBD and MSA and to discuss ways the Federal Government might help.
The Michael J. Fox Foundation is certainly aware of PSP and this year is co-funding multiple research projects in PSP alongside CurePSP. Its website mentions PSP as a condition related to PD that needs to be considered as a diagnostic possibility alongside PD.
Celebrities, certainly not exempt from PSP, have stepped forward:
In 2020 and 2021, “Zoey’s Extraordinary Playlist” was a series on NBC. One of its producers had a relative with PSP and arranged for the main character’s father, played by Peter Gallagher, to be similarly affected. The actor did a reasonable job mimicking its disabilities and the effects on his family were portrayed in an accurate, thorough and sympathetic way.
Linda Ronstadt announced that she has PSP in 2019, six years after receiving an initial diagnosis of PD. The long survival and initial misdiagnosis as PD suggest that she has the “PSP-Parkinsonism” subtype, but I have no inside information.
Back in 1999, the British-American comic actor and musician Dudley Moore was diagnosed with PSP. A few months before he passed away in 2002, he and his family organized a high-profile fundraiser for CurePSP at Carnegie Hall in New York City.
In 2023, Congresswoman Jennifer Wexton of Virginia announced that she had PSP. She retired in 2024, but not before using an AI-powered speech enhancement device to deliver a speech on the House floor. She was instrumental in adding PSP, CBD and MSA to a bipartisan bill establishing a way to efficiently coordinate all Federally-supported research in that area. The bill easily passed and is now the Dr. Emmanuel Bilirakis and Honorable Jennifer Wexton National Plan to End Parkinson’s Act.
Rev. Jesse Jackson, the 84-year-old civil rights leader and activist, announced his diagnosis of PSP in 2025.
I have no doubt that once a drug shows promise against PSP and looks like it will be approved, the sponsoring pharma company will cover the Earth with PSP awareness. Let’s all hope that happens soon, but until then, anyone can help. Just mention something about PSP on social media platforms and other Websites. Include links to CurePSP (psp.org) and to other reputable sources of information and support (hint – hint).
In a reply to a commenter back in July 2022, I expressed pessimism about the neuroprotective potential of buntanetap, formerly known as posiphen. That drug is claimed to reduce genetic transcription of multiple neurodegeneration (NDD)-related proteins, including tau, at the messenger RNA (mRNA) level. I thought it unlikely that one drug could do that without impairing production of other proteins necessary to health and life.
Later in the same year, 2022, a double-blind trial of buntanetap in Parkinson’s disease and Alzheimer’s disease was published. The N was small, with 54 participants with PD and 14 with AD. The treatment period was very short, only 25 days, which means that any result would reflect only symptomatic, not neuroprotective (disease-slowing), effects. That’s because the progression of PD and AD are too slow to be measured over only a 25-day period. However, the main purpose of the trial was to study safety and tolerability, not efficacy. The trial also checked spinal fluid levels of multiple proteins associated with the diseases – i.e., biomarkers.
The result were small but statistically significant improvement in some of the cognitive and motor measures in PD and some of cognitive measures in AD. Most of the biomarkers showed improvement, but none were statistically significant.
Some cell/molecular biology here, folks: The authors argue that previous attempts to treat NDDs by targeting only one protein at a time have failed because all the diseases actually include defects in multiple proteins. They point out that the mRNAs of multiple NDD-related proteins and iron-metabolizing proteins share a section called an iron-responsive element (IRE). But the mRNA for the iron-metabolizing proteins is very slightly different from that for the NDDs. IREs are not translated into any part of its parent protein, but they do regulate that protein’s production by the ribosomes: When the level of iron in the cell is too high, the IRE prevents the ribosomes from making the protein. The butanetap molecule binds to a specific part of the IRE, mimicking a high-iron effect. Luckily, the drug does not bind to mRNA of actual iron-metabolizing proteins – only to that of NDD-related proteins.
So, if you skipped that nerd interlude, the executive summary is that there’s a good scientific rationale for why buntanetap could work.
But there are some potential issues with the 2022 clinical trial:
Many of the patients experienced minor side effects such as headache, rash or muscle spasms. This could have impaired the blinding scheme and produced a placebo effect. That issue could have been addressed by asking the patients at study’s end, before the randomization assignments were revealed, whether they thought they had been on placebo or active drug.
Some of the spinal fluid markers that improved related to neuro-inflammation.That, and the improvement in the NDD-related markers, would not have been directly caused by a placebo effect. However, the increased mobility resulting from a placebo effect might have reduced neuro-inflammation, reducing in turn the markers of brain tissue damage. (Only the patients with PD, not AD, received testing for physical mobility and there was no record of the patients’ exercise habits.)
Like many drug trials at this early stage, this one was not only sponsored by the drug company, but the first and last authors were company employees. I found no evidence of dishonesty in reading the paper, but a potential financial and professional conflict-of-interest does exist.
As stated, this trial was only 25 days long, and we really need to know the longer-term side effects of depriving the brain of a whole raft of normal proteins, even if some of those molecules later cause mischief.
Enough for now. My next post will discuss the results of a larger, more recent trial of buntanetap that has not yet been published in a peer-reviewed journal. Cliffhanger.
My post on June 24, 2025 was entitled, “I like Parkinson-like.” It was about a paper that was about to hit the stands (a Boomerism) authored by myself and Dr. Junaid Siddiqui of Cleveland Clinic. It proposed replacing the terms “atypical Parkinsonism,” “atypical Parkinsonian disorder” and “Parkinson-plus disorder” with a new term, “Parkinson-like disorder” (or “Parkinson’s disease-like disorder” because let’s not get TOO prescriptive).
The June blog post promised to make a copy of the paper available to you all once it was published. Better late (by seven months) than never. My mind was jogged by one of my current projects, preparing a lecture on the atypical Parkinsonian disorders in general that I will deliver at the University of Vermont in April. Also jogging my mind was that it’s been nearly two weeks since my last post.
The pdf is at the bottom. If you have difficulty downloading or viewing it, here’s a link to the abstract and highlights and here’s a chart showing the gist of our proposed new terminology:
Yesterday’s post was about how the 10 most-frequently-visited news items in Parkinson’s News Today (PNT) of 2025 might relate to PSP. The list appeared here.
#5: FDA clears early trial of stem cell therapy for Parkinson’s
The treatment, called XS-411, is made from stem cells derived from multiple healthy donors and converted into dopamine-producing cells. Each patient would receive one injection of such cells into each side of the putamen, the brain area where the dopamine synapses are located. The FDA approved a Phase I trial in April 2025, but now, nine months later, clinicaltrials.gov lists two small studies in China but none in the US. Most current stem cell trials in neurology derive the cells from the patient’s own bone marrow or skin biopsy, so I really don’t know why the company, XellSmart Bop-Pharmaceutical Company of Suzhow, China, is using cells from people other than the patients themselves. Maybe they have a new way to suppress the immunologic rejection or maybe production using multiple donors is more easily scalable to serve large numbers of patients. Could XS-411 work in PSP? Perhaps it could help the same fraction who respond to levodopa, which is only a minority, and their benefit is usually modest and transient. The problem is that in PSP, unlike in PD, the cells in the putamen receiving the dopamine-encoded signals are degenerating along with the dopamine-producing cells. So, we need more research into injecting stem cells producing neuroprotective molecules or non-dopamine neurotransmitters.
#4: Program offers psychedelics as treatment for Parkinson’s
Ibogaine is a psychedelic drug legal only in a few states, and even then only for FDA-approved research use. Ambio is a company offering the drug to people with a wide variety of conditions on a “research” basis at clinics in Mexico and Malta. There’s no such listing in clinicaltrials.gov, so I can’t be sure of the protocol except that patients are charged $6,050 for a four-day treatment initiation at one of their clinics and “micro-doses” for the next six months. Based on the little information available, this fits the profile of many “research trials” of alternative treatments: a hefty fee, minimal pre-treatment evaluation or followup, no control group and no peer-reviewed publication. In this case, I must also wonder about the risk of habituation to the treatment itself, and where do you suppose you could buy something to satisfy that? Enough – you now know what I think about this drug for PSP, even if the initial $6,050 and the travel expenses are not an issue for you. To be sure, some alternative treatments do have legitimate potential, but when there’s a major risk of financial or medical harm (both of which apply here), their use should be confined to formal FDA-approved research protocols.
#3: Bacteria in digestive tract tied to cognitive decline
Parkinson’s disease is a natural candidate for causation by intestinal bacteria because the first stage of the disease, aggregates of the alpha-synuclein protein in neurons, starts not in the brain, but in the intestines and lungs. But PSP does appear to start in the brain. There’s been little research on gut bacteria and PSP, but something important was reported in 2023 from researchers in China and summarized in my 4/2/23 post. That short-term study found that replacing the colonic bacteria produced about a 10 percent benefit in the PSP Rating Scale score. The trial was too short and small to conclude anything about long-term slowing of progression. Bottom line: Although PSP does not start in the GI tract as PD does, gut bacteria may play a role and should be studied further for any therapeutic implications.
#2: Study identifies potential way to treat Parkinson’s constipation
Ghrelin is a string of 28 amino acids with many basic gastrointestinal functions including stimulating appetite at the level of the brain and defecation at the level of the spinal cord. The article reported by PNT teased out an important detail that could hold implications for treatment of constipation in those with PD. We don’t know if applies as well to the constipation of PSP, but I can say that some of PSP’s constipation is caused by degeneration of a cluster of cells in the lower spinal cord that are not involved in PD. One research study, from 2013, did find a role of ghrelin in multiple system atrophy but not in PSP or corticobasal syndrome. So, those few strands of evidence suggest that people with PSP will have to rely on traditional methods of keeping things moving – exercise, fluids, fiber, a stool softener, and avoidance of drugs that block acetylcholine synapses (“anticholinergics”). Many drugs in the last category are used for other PSP symptoms such as imbalance, vertigo, urinary incontinence and depression, so getting off those is a good topic for discussion at the neurologist’s office.
#1: Research shows disrupted mitochondrial DNA tied to inflammation
For decades, we’ve known that in both PD and PSP, the cells’ mitochondria malfunction and there’s excessive inflammation in the brain. But we don’t know which is cause and which is effect, or if they’re both effects of a common cause. The new study used a novel genetic technique to find evidence that it’s the inflammation causing the mitochondrial loss. In theory, the same study could be performed in PSP. A similar result would suggest that to slow PSP progression, targeting excessive inflammation might do better than those targeting mitochondria directly. Early in my career, I had narrowed my subspecialty choices down to movement disorders and neuroimmunology/multiple sclerosis. I chose the former because I was flummoxed by the complexity of the immune system. Little did I know . . .