Now that I’ve shared the details of a work week and my breakfast menu, I’ll share my innermost thoughts. Or maybe not my innermost — just some neurology-related thoughts that have been occupying a lot of my conscious moments lately. No, these thoughts aren’t about anything very interesting, unless you have an atypical Parkinsonian disorder (APD), want the best possible care and want it fast.
Here’s the background: As CurePSP’s Chief Clinical Officer, I serve on the Steering Committee of CurePSP’s Centers of Care (CoC) network. That’s a group of 28 movement disorders referral centers in the US and 2 in Canada with particular interest and qualification in the care of PSP, CBD and in many cases, MSA. (The centers are already in place – CurePSP does not create or operate them, but it does provide each CoC a token $5,000 per year to help defray expenses.) The network’s mission is to improve the quality of, and access to, first-rate care for these diseases, both at our own centers and generally. In 2021, the group published a “best practices” document on the symptomatic management of PSP and CBD. (That means how to treat the symptoms to make patients’ lives better until we actually have a cure.)
One of the current goals of the CoCs is to make it easier for people to be evaluated by a doctor with the training and experience needed to tell them if they have PSP, CBD or MSA, and if not, what they do have, and then to advise them on prognosis and management. One way the CoCs try to do that is to expand our ranks until we have a center within a reasonable drive of most of the population. Another is to reduce the wait for appointments, which among the CoCs averages 3½ months and for 14% of the CoCs, exceeds 6 months.
Why is 3½ months (or 6!) too long? Because PSP, CBD and MSA can progress quickly. The average patient with these diseases survives only about 3-4 years after receiving the correct diagnosis, so 3½ months is a big chunk of that. Could an oncologist tell a woman with an abnormal mammogram to wait 3½ months to be seen? No, and they don’t. Even (or especially) the busiest cancer specialists have figured out how to see new patients within a few days.
We would like the wait for an initial appointment for suspected APDs not to exceed one month. One of the ideas the CoCs have been batting ideas around in our Zoom calls is to reserve a couple of hours each week just for patients with known or suspected APDs and then, to prevent those time slots from being overwhelmed, to reduce demand. In other words, provide referring physicians – usually general neurologists – with a convenient diagnostic algorithm for the APDs to allow them to chose the next diagnostic tests themselves and initiate management. Then, a confirmatory evaluation with the subspecialist can wait 3½ months without much risk of harm.
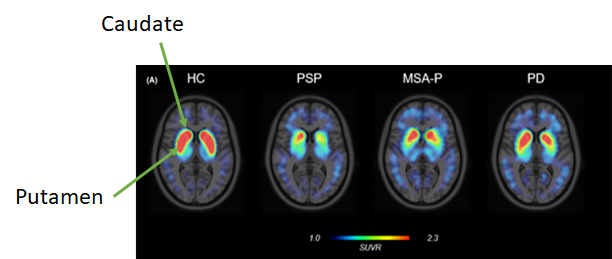
To that end, the CoCs are working on a practical guide for general neurologists on how to diagnose all the APDs, and I do mean all – not just to recognize PSP, CBD and MSA, but also to recognize 49 other progressive disorders in adults that can mimic aspects of PSP, CBD and MSA.
It’s hard to get busy physicians to sit down and read a textbook, especially about diseases they’re not going to see very often. So, we’re describing a step-by-step process in the form of a few charts to allow general neurologists to apply a diagnostic decision tree in real time to 55 possible disorders and initiate treatment. We’re deep into the process as a group, so I can’t say more right now, but I will as soon as I can.
What I can say now is that the final product will be available for free on the CurePSP website and by a link from this blog.