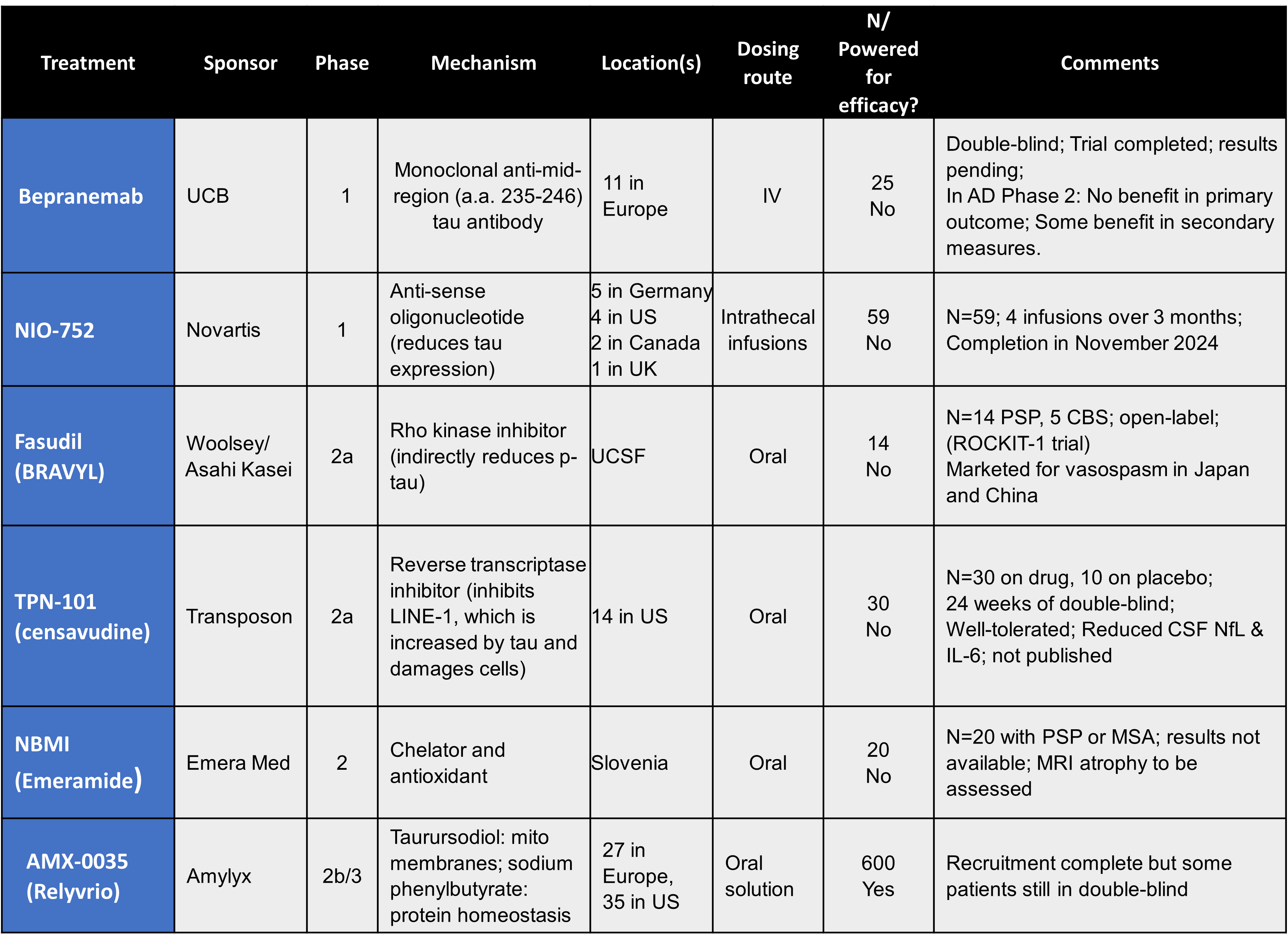
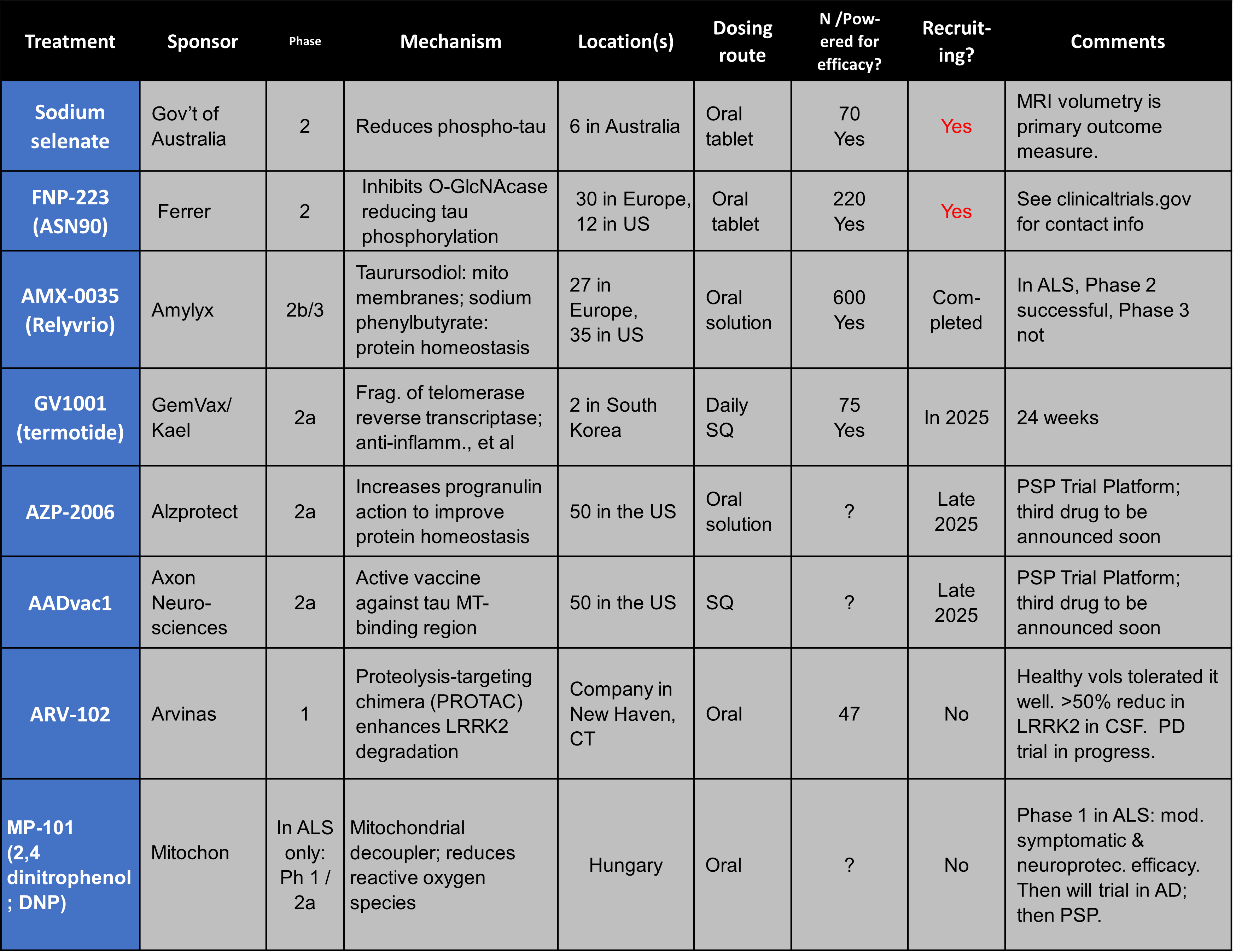
A new candidate has emerged in the ever-frustrating quest for a valid diagnostic test for PSP. It turns out that microRNA in tear fluid has something to contribute.
As you no doubt recall from high school biology, DNA is transcribed in the cell’s nucleus into long strands of messenger RNA, which are then translated by the ribosomes into proteins. The messenger RNA strands can be hundreds of nucleotides long. (Nucleotides are the four “letters” comprising the genetic code.)
But some genes, or parts of genes, are transcribed into RNA that’s never translated into protein. These short stretches, called microRNA (miRNA), have only 21 to 23 nucleotides. They act on regular messenger RNA in some way to reduce its likelihood of being translated into its protein. Essentially, this system is a way for some genes to regulate others. About 60% of our 20,000 genes are regulated by miRNAs and errors in this molecular mechanism are associated with (but not necessarily causing) things like Parkinson’s, Alzheimer’s, Huntington’s, cancers of various kinds, atherosclerosis, kidney diseases, obesity, clotting disorders and alcoholism.
It turns out that sometimes miRNA leaks from the interior of nerve, brain or spinal cord cells into accessible body fluids like blood, spinal fluid and urine, where it can be sampled and tested using PCR, that now-familiar test for diagnosing Covid and catching crooks.
It’s pretty hard to sample spinal fluid, but blood is a lot easier, and urine is easier than that. Easier still is tear fluid. All it takes is a quarter-inch-wide strip of filter paper, like that in coffee filters, bent into a hook that hangs from the lower lid for 5 minutes, absorbing tear fluid.
A new publication in the journal Molecular Neurobiology from Dr. Antonia Demleitner and colleagues in Munich, Germany have taken the first step to using tests of miRNA in tear fluid to differentiate among PSP, MSA and PD. The study was small, with only 10 participants with PSP, 29 with PD and 7 with MSA, plus 10 healthy people as controls. In each of the 56 people in the 4 groups, they measured levels of 1,113 kinds of miRNA.
After correcting the data for things like age, disease duration and medications that might reduce tear production, they found 286 miRNAs in all four groups and 244 in none. 35 kinds of miRNA occurred in at least some members of the PSP group and in no one in the other three. 27 miRNAs occurred in all groups except PSP.
They then uploaded their data into an app that finds commonalities among groups of miRNAs. It found that the miRNAs present exclusively in PSP had to do with immune function and inflammation, the apoptotic pathway (a normal program by which cells cause their own death when necessary), and the microtubules (the brain cells’ internal skeleton/monorail system that the tau protein helps maintain).

The miRNAs absent in PSP but present in the other 3 groups tended to be associated with the function of a certain trophic factor (a protein devoted to helping brain cells sprout new connections and maintain the old ones).
If a tear-based test for PSP becomes available in a few years, it could make it much easier to know at an early stage of the illness if some new PSP medication would be worth trying. It would also make it easier to recruit patients into clinical trials hoping to find such medications. Non-interventional research such as surveys of environmental risk factors would also be easier to do and more valid with a diagnostic test that works in early stages of the disease and is cheap, harmless and painless. Even if miRNAs provide a useful diagnostic test, it doesn’t mean that the genes they regulate are the causes of PSP. Equally, or more likely is that those miRNAs are produced as a normal response to the damage being caused by the PSP process.
As Dickens indicates in the above quote from Great Expectations, tears can actually solve problems.