Back in 2023, I posted an explanation of the ten PSP subtypes. The archetypal subtype, PSP-Richardson syndrome accounts for about half of all PSP and, in contrast to most of the other subtypes, has a rapid progression rate, a validated rating scale, and highly accurate diagnostic criteria. All of these features have led clinical trial sponsors to maximize their trials’ sensitivity and minimize their costs by restricting admission to people with PSP-Richardson. But developing better outcome measures for non-Richardson forms of PSP could change that practice.
A big step toward realizing this goal was published last week in the journal Neurology by a group at the Mayo Clinic in Rochester, MN. Led by first author Dr. Mahesh Kumar and senior authors Drs. Jennifer Whitwell and Keith Josephs, the study found that a good outcome measure for clinical neuroprotection trials in all PSP subtypes was to combine a measure of atrophy by MRI with a measure of clinical disability. This is a major advance.
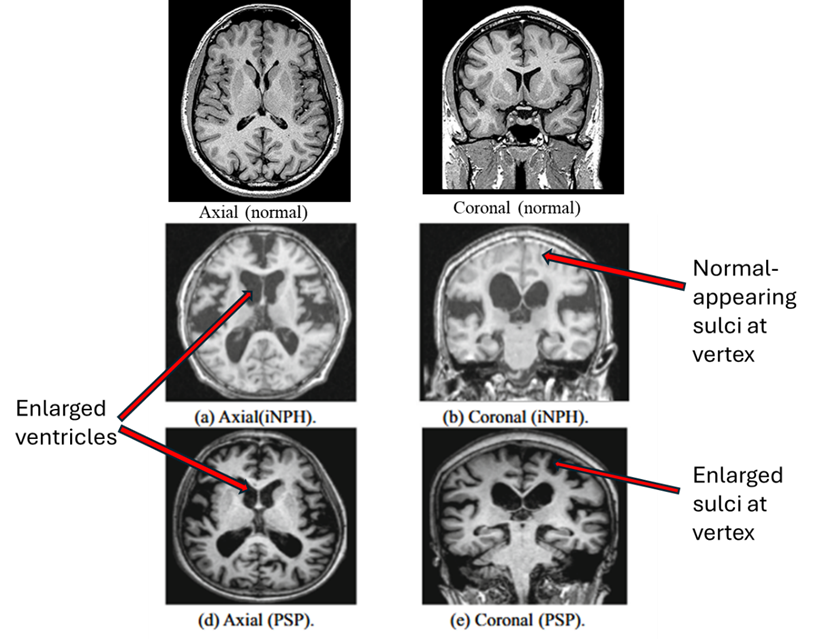
The researchers performed brain MRIs at the start and end of a one-year period in 88 people with PSP and 32 age-matched controls. Of those with PSP, 50 had PSP-Richardson, 18 had “PSP-cortical” (three of the other nine subtypes) and 20 had “PSP-subcortical” (the other 6 of the subtypes). They had to lump the non-Richardson subjects using their subtypes’ general anatomical predilections because most of the subtypes were too rare to analyze on their own.
Calculating how much each of ten important PSP-involved brain regions had atrophied over the one-year interval allowed the researchers to identify which region(s) might best serve as markers of progression for each of the three groups when coupled with standard clinical measures. Those measures include such familiar instruments as the PSP Rating Scale and the Unified Parkinson’s Disability Rating Scale’s motor section as well as less familiar scales specific for cognition, gait, eye movement and speech. All the scales were administered concurrently with each of the two MRIs.
They expressed the sensitivity to one-year progression not by some abstract statistic, but by the number of patients needed in a double-blind trial to demonstrate with at least 80% certainty that patients on active drug enjoyed a 20% slowing of progression relative to the placebo group. (These specifications are typical for PSP clinical trials.) The better the measure’s performance, the fewer patients are needed.
And the award for Best Performance by an Outcome Measure in a PSP Neuroprotection Trial goes to . . . a combination of the rate of atrophy of whatever brain region shrinks fastest in the patient’s specific subtype and the PSP Rating Scale score.
The real significance of this study’s result is that using an outcome measure customized to each participant’s PSP subtype could allow trials to enroll not just people with PSP-Richardson, but also those with any of the other subtypes. That’s because the trial’s measure of success could be to compare each patient’s rate of progression during the trial to that of patients in the placebo group with the same PSP subtype.
This could double the number of people eligible to enroll in PSP trials, which means cutting the enrollment period in half, with commensurate reduction in costs for the sponsor. The hybrid measure is more sensitive to progression than the PSP Rating Scale alone, thereby reducing the number of patients required even more.
Both factors could lower the financial barrier confronting a company hoping to mount a trial for a promising PSP drug. That may be the most important bottleneck right now in the development of a treatment to prevent or slow the progression of PSP.s
That’s why this news is huge for PSP in general and for the “orphans” in particular.