Today the Spanish drug company Ferrer issued a press release announcing the successful completion of enrollment in the PROSPER study. That’s the year-long, double-blind trial of FNP-223 that I’ve told you about in September 2025, June 2025, October 2024 and April 2024. The mechanism of action is to prevent phosphate groups from being attached to the tau protein.
The recruitment required only 11 months, one month less than planned. Now, the last-enrolled patient will require 12 months to complete the trial and then the data will take a few weeks to be “cleaned.” (That sounds like scientific hanky-panky, but actually it means tracking down records for missing test results, resolving contradictory information, and getting signatures from all the neurologists on everything.) Then the statisticians take a couple of months to do their thing, producing a result. So, we’re talking early 2027.
I haven’t a clue as to whether FNP-223 is likely to work in slowing the progression of PSP. I do know that its oral administration is a plus and its mechanism of action at the subcellular level makes sense . I also know for sure that hope matters!
[Disclosure: I consulted for Ferrer in their trial design and implementation, but I have no financial stake in the trial’s outcome or the company’s success.]
Some excellent news for you today. The orally administered drug AZP-2006 has shown early signs of slowing the progression of PSP. (Yes, you heard right!)
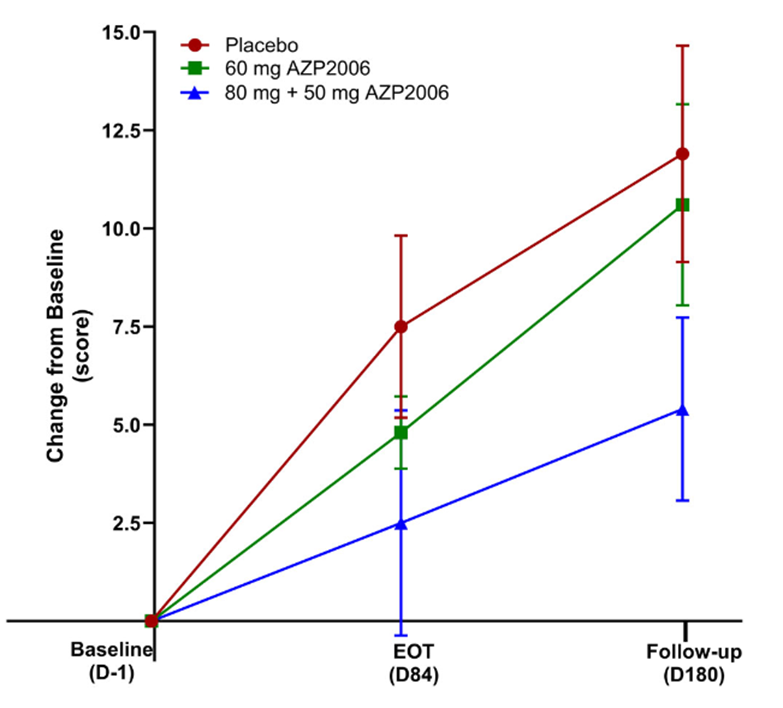
My blog post from May 9 of this year brought news that a small, open-label, Phase 1 study of AZP-2006 seemed to have slowed the progression of PSP by 31 percent. Now, the drug has completed a small, double-blind, Phase 2a trial with even better results: In the 11 patients receiving 60 mg per day, the worsening in the PSP Rating Scale score over the 3 months of the double-blind phase was a third slower than in the placebo group (identical to the result of the uncontrolled Phase 1) and in the 13 patients receiving a loading dose of 80 mg on the first day and then 50 mg per day, the apparent worsening was two-thirds slower.
It’s important for you to understand, and the authors repeatedly emphasize, that these results were not statistically significant, meaning that they could be the result of a random fluke. There were also some minor differences among the three patient groups (placebo, 60 mg, and 80 mg then 50 mg) at the study’s baseline that theoretically could have explained the results. A larger, Phase 2b study could confirm the result while having the statistical power needed to compensate for any “baseline bias” among the treatment groups.
The trial included a 3-month open-label extension. That’s where the participants on placebo for the first 3 months were offered the opportunity to convert to the active drug at 60 mg per day, while those initially on the active drug could opt to continue it. Over months 4, 5 and 6, the rate of decline of the formerly-placebo group slowed down noticeably. The other important result is that the drug showed itself to be safe and well-tolerated over the entire 6 months.
The publication’s first author is Jean-Christophe Corvol, MD, PhD, a very well-regarded, senior neurologist I know at the legendary Hôpital Pitié-Salpêtrière in Paris. The senior (i.e., last-named) author is Luc Defebvre, MD, PhD, at Lille University. Six of the other 16 authors are staff researchers at the sponsoring drug company, AlzProtect, of Lille, France.
In this graph, the vertical axis is the worsening in terms of the 100-point PSP Rating Scale. EOT is end of the double-blind part of the trial at Day 84. Thereafter, all participants received active AZP-2006. Note that both active-drug groups progressed more slowly than the placebo group over the first 3 months; and on active drug, the participants formerly on placebo may have slowed their progression rate. The vertical line segments represent standard deviations of the mean. (From: Corvol JC, Obadia MA, Moreau C, et al. AZP2006 in progressive supranuclear palsy: outcomes from a Phase 2a multicenter, randomized trial, and open-label extension on safety, biomarkers, and disease progression. Movement Disorders. 2025 Sep 27. doi: 10.1002/mds.70049. PMID: 41014124)
So, when will the Phase 2b study start? My May 5, 2025 post reported on the “PSP Platform Trial,” (PTP) an NIH-supported collaboration among dozens of U.S. academic centers to perform Phase 2b trials on up to three drugs simultaneously using one placebo group. One of the first three drugs, in fact, is AZP-2006. Last I knew, the PTP was expected to start late this year, but it’s now almost October and I’ve heard nothing further other than that some details remained to be ironed out with the FDA. That trial would take about 6-12 months to recruit and then another 12 months for the last patient to finish, then at least a couple of months to analyze the data.
So, how does AZP-2006 work? I’ll plagiarize my own May 9 blog post, along with its “Nerd Alert!” warning that this gets technical:
The main mechanism of action of AZP-2006 is at the lysosomes, one of the cell’s garbage disposal mechanisms, where it acts specifically at the lysosome’s prosaposin and progranulin pathways. Prosaposin is the metabolic precursor (a “parent molecule” cleaved by enzymes to produce the active molecule) of the saposins, a group of proteins required for the normal breakdown of various types of lipids that are worn out or over-produced or defective from the start. Progranulin is the precursor, as you’d guess, of granulin, which, like saposin, is involved in function of the lysosomes. But progranulin addresses disposal of proteins, not lipids. In mouse experiments, the drug also enhances the production of progranulin, mitigates the abnormal inflammatory activity in tauopathy, reduces tau aggregation, and stimulates the growth or maintenance brain cell connections.
Bottom line: This very small, Phase 2a trial was designed to show safety, not efficacy, and its slowing of PSP progression did not nearly achieve statistical significance nor exclude potential sources of random bias. But the magnitude of the (apparent) effect make this excellent news for those with PSP, present and future.
In case you missed the front-page story three days ago (3/8/24) in The New York Times, the drug Relyvrio has failed to show benefit in a large (664 participants), Phase 3 trial in amyotrophic lateral sclerosis. The drug did appear to show benefit in a much smaller (137 participants) Phase 2 ALS trial in 2020 and was provisionally approved for sale by the FDA on the strength of that result. Now, the drug company, Amylyx, may have to discontinue marketing the drug for ALS.
Why is this relevant to PSP? Because four months ago Amylyx initiated a 600-participant Phase 3 trial of Relyvrio for PSP. It’s called “ORION.” So far, recruitment has begun at only a handful of sites, all in the US, with plans to expand into Europe and Japan over the coming months. The FDA’s permission to start ORION without a Phase 1 or 2 in PSP was based in part on the success of the drug in the Phase 2 ALS trial. See my post of February 29 for details.
Amylyx is also testing Relyvrio in people with Alzheimer’s disease, where a Phase 2 trial has demonstrated adequate safety and tolerability. I have no information on a Phase 3 in AD.
The question now is whether the ORION trial in PSP will continue. So far, there’s been only one business day since the ALS news, and I’m not sure if the top brass at Amylyx — or the company’s sources of financing — have yet decided. But the Times article reported that the FDA approved Relyvrio for ALS only after Amylyx agreed to withdraw the drug if the Phase 3 trial showed no benefit. Furthermore, right after the ALS trial news hit, the stock price of Amylyx dropped from $19 to $3 and stayed about there. Stock markets usually know how this sort of news is likely to play out. Relyvrio is Amylyx’s only marketed product but they do have other drugs in the development pipeline.
The mechanism of action of Relyvrio addresses issues important to both disorders, which suggests that if it failed in one, it could well fail in the other. But we don’t really understand the pathogenesis of either disease well enough to know if PSP might respond when ALS did not.
Meanwhile, if you were planning to try to enroll in the ORION trial, I’d advise you not to change your plans. The ALS trial showed no important toxicity, at least in people with ALS, and you wouldn’t want to lose your potential spot at the study site because you delayed enrolling until definite information on the future of the ORION trial became available. When other trials in PSP start enrolling, that advice could change, of course.
(I’ll repeat the disclosure I made in my 3/8/24 post: I’m a paid consultant for Amylyx, advising them on design of the ORION trial and training the participating neurologists on proper administration of the PSP Rating Scale. But I have no stock in the company nor other financial interest in the drug’s success.)
I hope you will forgive my 24-day posting hiatus. To make it up to you, I bring good news: The trial of AMX-0035 in PSP is planning to expand its enrollment activities in the next few weeks, and this drug’s track record is unusually encouraging.
The trial, dubbed “ORION” for some reason, initiated enrollment over the past two months at eight sites in California, Florida, Massachusetts, Michigan, Tennessee and Texas. 32 other sites in the US and dozens in Europe and Japan will open in coming months, with a total enrollment target of 600 patients. Those interested can email clinicaltrials@amylyx.com, check clinicaltrials.gov or the company’s own site. The trial will include a 12-month double-blind period with a 40% chance of assignment to the placebo group, followed by a 12-month open-label period. Trials like this usually take about a year or two to fully enroll, another year for the last enrolled participant to complete the double-blind and another few months to analyze the data.
The drug company is Amylyx Pharmaceuticals, based in Cambridge, Massachusetts. They held a meeting a few days ago for their US sites’ neurologists and coordinators, where I gave a detailed lesson on proper administration of the PSP Rating Scale, which will be the study’s main outcome measure. (Disclosure: Amylyx paid me for that presentation and for general advice on the trial’s design but I have no financial interest in the success of the company or the drug.)
The treatment in question is actually two drugs, taurursodiol and sodium phenylbutyrate, both administered orally as a powder stirred into water. The first addresses the dysfunction of the mitochondria in PSP. The second reduces stress in the endoplasmic reticulum and enhances the unfolded protein response, both of which are also dysfunctional in PSP. All of these cellular functions are related and lab experiments show that the two drugs combined work better than the sum of their individual effects.
Unlike any of the other new drugs currently or or soon to be tested for PSP, AMX-0035 has been found to help a related disease, amyotrophic lateral sclerosis (ALS or Lou Gehrig disease), where it appears to slow the progression by about 25% and prolongs survival accordingly. The drug, branded “Relyvrio,” won approval from the FDA for ALS last year and is gaining widespread acceptance among neurologists in treating that condition.
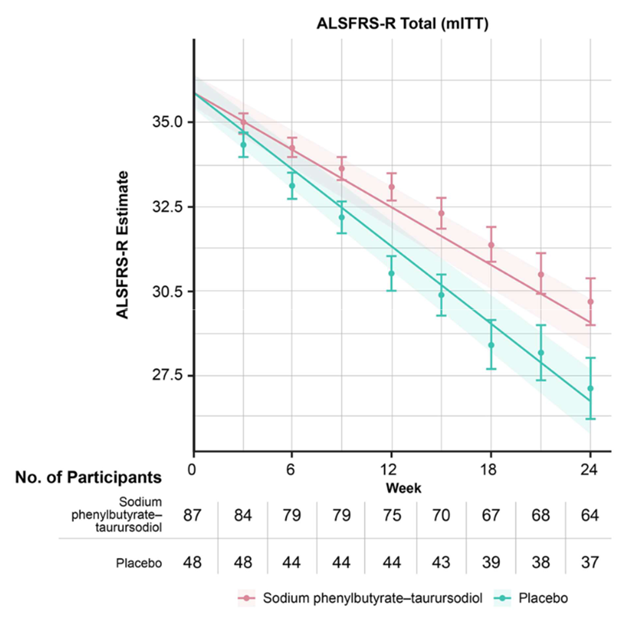
The graph below (from Paganoni et al, New England Journal of Medicine, 2020) shows the worsening of the main ALS disability measure (vertical axis; note that the bottom is not zero) over the 24 weeks of the trial (horizontal axis). The orange line/shaded areas and the means/standard error bars represent the patients on AMX-0035 using two different statistical techniques. The patients on placebo are shown in green.
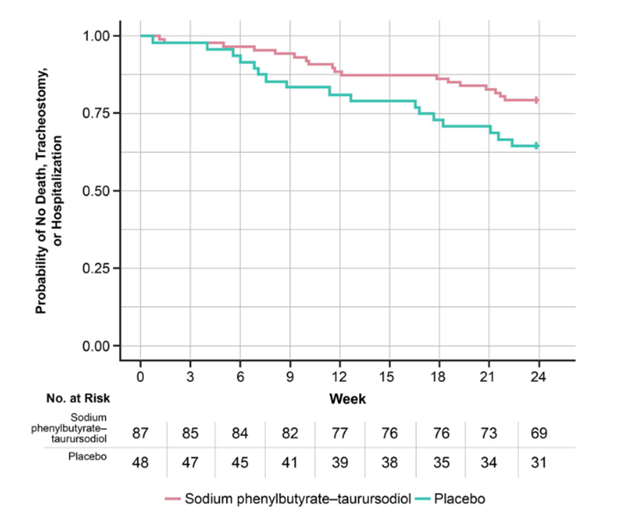
The graph below (also from Paganoni et al), called a Kaplan-Meier survival plot, shows the fraction of patients in the ALS trial remaining alive without tracheostomy or hospitalization (vertical axis) along the 24 weeks of the trial (horizontal axis).
This is great for people with ALS, but that’s not a tau-based disorder like PSP. However, in a Phase 2 trial in Alzheimer’s disease, which is partly a tau disorder, AMX-0035 did reduce spinal fluid levels of both total tau and of a toxic form called p-tau 181. That trial was too small and brief to reveal any efficacy of AMX-0035 to slow or halt AD progression but I assume a proper Phase 3 trial will follow.
Side effects of AMX-0035 in the AD trial have not been published, but in the ALS trial, nausea, diarrhea, excess salivation, fatigue and dizziness occurred in 10% to 21% of patients on the drug and in slightly lesser percentages of those on placebo.
If AMX-0035 shows the same result in PSP as it did in ALS, that means about one additional year of survival for the average patient, and even more if the disease can be diagnosed earlier. Potential game-changer. I’ll keep you updated.
With a nice handful of medications for PSP approaching clinical trials, it would be great to be able to assess the participants’ movement ability not just every few weeks to months at the research center, but also much more frequently at home. The reduced need for clinic visits would ease participation for patients who for whatever reason have difficulty tolerating or obtaining travel. It could also provide a more “real-world” picture of how the patient is doing in their home environment.
One relatively easy step in that direction arose from a project published last year (in which I, full disclosure, was senior author). It modified the 28-item PSP Rating Scale, omitting the exam items that might not work well by video and used existing databases of PSPRS scores over time to assess the correlation between the modified and unmodified scores. In short, the correlation was excellent.
But a PSP Rating Scale modified for video still requires a video connection, and that can be tough for the PSP age group and their caregivers, especially those where cell service is spotty. Besides, video visits can’t happen every day or even close to it. So, some other gadget would be nice.
Now, a group led by Dr. Alexander Pantelyat of Johns Hopkins and Dr. Anne-Marie Wills of Mass General (the co-senior authors) with Dr. Mansi Sharma of Mass General (the first author) have published a first-blush look at a simple gait monitoring system in PSP and Parkinson’s.
Other versions of the same idea for PSP have had to be used in a lab at a research facility and required a complex array of sensors pasted to various parts of the body. But this one is used in the patient’s home and requires only three sensors: one strapped to the lower back with a belt and one fastened to each shin by what looks like an old-fashioned garter strap like my father used to wear. For reasons of safety, only patients with histories of very few falls and ability to walk unassisted qualified for this early trial. Patients with PSP and Parkinson’s were compared on their performance of four standard gait tasks. They received instructional videos and the three sensors communicated with an app on a tablet provided.
Of the 22 patients who qualified and consented, only two (both with Parkinson’s) couldn’t manage the technical requirements. For the others (10 with PD and 10 with PSP), the device proved able to quantify and time the movements well and to differentiate PSP from Parkinson’s. Most important was that managing the experimental hardware and software while avoiding falls or other complications was perfect.
The next step will be to assess the device over a period of several months for its ability to track PSP progression. This should be successful because the Spearman correlation coefficients of the three gait measures with the modified PSP Rating Scale, were pretty good: 0.62, 0.64 and 0.84; and we know that the PSPRS tracks PSP progression well. (Correlation of 1.0 is perfect and 0 is random.)
Another reason to be optimistic about the device to track progression is that it’s already been accomplished, although with a more complex, six-electrode device implemented in a research lab.
A reason for caution is that not every patient in a drug study walks unassisted at home as safely as these 20 hand-picked participants, especially toward the end of a one-year trial period. Furthermore, using this device at home in routine clinical practice would involve patients at all levels of gait instability. But for people in remote areas or whose caregiver can’t afford to take time off from work for a clinic visit, this could be the ticket to research trial participation.
One of this blog’s more frequent and thoughtful readers/commenters, “Mauraelisabeth3,” has asked a good question about the possibility auditory and visual sensory gamma-frequency stimulation as a treatment for PSP. I responded by promising a blog post on the subject, and here it is:
As always, some scientific background first: An electroencephalogram (EEG) is a recording of electrical waves emanating from the surface of the brain, as measured by wires pasted to the scalp. The various brain waves are classified by their frequencies. The ones relevant to ordinary patient care range from the slowest (i.e., lowest frequency), called “delta,” at 1-4 cycles per second (or “Hertz” or “Hz), to “theta” (4-8 Hz), “alpha” (8-13 Hz) and “beta” (13-30 Hz). Most of the EEG activity in a healthy, relaxed but awake adult with eyes closed is alpha, and with increasing alertness or with eye opening, there’s more beta. Theta and delta are important in normal sleep and in many kinds of brain diseases.
But there’s a higher frequency called “gamma,” which can be found mainly in deeper areas of the brain not usually detected by routine EEG, or if it is detected, it’s hard to distinguish from artifact caused by scalp muscle activity. It turns out that in people with Alzheimer’s disease, there’s a reduction in gamma activity in the areas deep in the brain that are the headquarters of the memory problem.
Now to the matter at hand: There’s a way to “entrain” the EEG activity of those memory-related areas to increase their gamma activity. An AD mouse model called 5XFAD (with 5 mutations in 2 genes relevant to AD: amyloid precursor protein and presenilin 1) improves in multiple ways after such stimulation, including reducing its load of beta-amyloid, the main component of the amyloid plaques of AD. A mouse with a mutated form of the tau protein shows improvement in some measures as well, and tau, of course, is the protein central to PSP. Here’s a technical review article on the topic, but it’s from 2018.
A company called Cognito Therapeutics, based in Cambridge, MA, was started by scientists at MIT who have performed much of the early lab work. So far, the company has sponsored one small, controlled trial showing some improvement in people with AD, but it’s not published other than in very cursory form on the company’s website. A year ago, in December 2022, Cognito started a Phase 3 trial in AD, meaning a large trial of the sort that, if successful, could win FDA approval for the device for AD. It’s scheduled to conclude in 2025. For one hour a day, participants wear glasses flashing a light at 40 Hz (the most relevant point in the gamma range) and headphones playing a tone at the same frequency. A 40 Hz flash is just barely perceptible as flashing (50 Hz is the standard “fusion frequency”) and a 40 Hz tone is a low rumble.
Some caveats about the treatment:
The most recent literature I found is far from unanimous on whether AD consistently has reduced gamma activity in relevant brain regions, and I found no evidence at all that PSP does.
Although the small clinical trial to date found no adverse effects, there is evidence from the mouse experiments that the gamma stimulation increases the activity of the microglia, the brain’s main inflammatory cells. That could be a good thing if it enhances the scavenging of unwanted, aggregation-prone protein. But it could be a bad thing if it aggravates the inflammation thought to comprise an important part of the pathogenetic process in many neurodegenerative diseases, including AD and PSP.
In the few clinical trials to date, an hour’s stimulation provides only about one day of measurable benefit. Such a regimen might prove impractical in the real world.
Bottom line: Would I recommend volunteering for a trial of 40-Hz sensory stimulation in PSP . . .
. . . if some more lab data in mice, or very early phase human data supported the benefit and safety of such a treatment in PSP, and
. . . if such a trial fully communicated the scientific uncertainties and safety concerns?
Yes, I probably would.
Keep in mind that devices producing 40-Hz light and/or sound are already commercially available as meditation aids. No clue here if they help, harm or neither, but until I know more, I’ll categorize them along with all the other placebos out there.
My periodic updates on active PSP neuroprotection treatment trials have mentioned a drug called TPN-101. That’s an oral drug that inhibits an enzyme called reverse transcriptase. If that term sounds familiar, it’s because that’s one of the mechanisms of anti-HIV drugs.
Transposon Therapeutics issued a press release yesterday announcing that a 24-week, Phase 2 trial of TPN-101 in 30 patients showed a reduction of spinal fluid levels of neurofilament light chain (NfL) by 18.4% compared with the 10 patients on placebo. NfL is a normal protein in brain cells that leaks out into the spinal fluid during active brain degeneration. TPN-101 also reduced spinal fluid levels of interleukin-6 (IL-6) by 51.6%. IL-6 is a component of the immune response in the brain that correlates with inflammation, part of the neurodegenerative process in PSP. There were no important side effects.
A study of only 30 patients is far too small to show any outward neuroprotective effect that might exist. This trial was designed to look for chemical evidence of engagement with the “target” cells and proteins in the brain and also to detect major side effects.
The findings will be presented as a poster at the 18th International Conference on Alzheimer’s and Parkinson’s Diseases in Lisbon in March 2024.
If you’d like to know how this drug works, put on your nerd hat and hang on: Our genomes are riddled with short stretches of DNA called “transposable nucleotide elements” inserted there by a viruses infecting ancestors hundreds of millions of years ago. But we have ways to prevent this viral DNA from being translated into proteins. One protective mechanism, called “chromatin packing,” uses a variety of proteins to surround our DNA strands like insulation on a copper wire, preventing our protein-making machinery from gaining access. The chromatin, however, does have to grant access to allow normal protein manufacture, and as we age, the chromatin starts to grant too much access. The old viral DNA can now be encoded into RNA, which our immune system promptly recognizes as foreign. The result is an inflammatory immune response that, via a variety of pathways, encourages the tau protein to misfold and aggregate. Those, of course, are the hallmarks of PSP and a couple of dozen other “tauopathies.” TPN-101 inhibits an enzyme called “LINE1 reverse transcriptase,” which is necessary for the transcription of the transposable nucleotide elements into RNAbut is not involved on normal cell processes. In other words, the drug puts that cat back into its bag.
I hope and assume that the next step will be a larger study attempting to show clinical benefit in slowing progression of PSP. This typically takes months to organize, months more to recruit all the patients, 12 months for the last-recruited patient to complete the double-blind phase, and another few months to analyze. That totals about 3 years, but at least things are moving in the right direction.