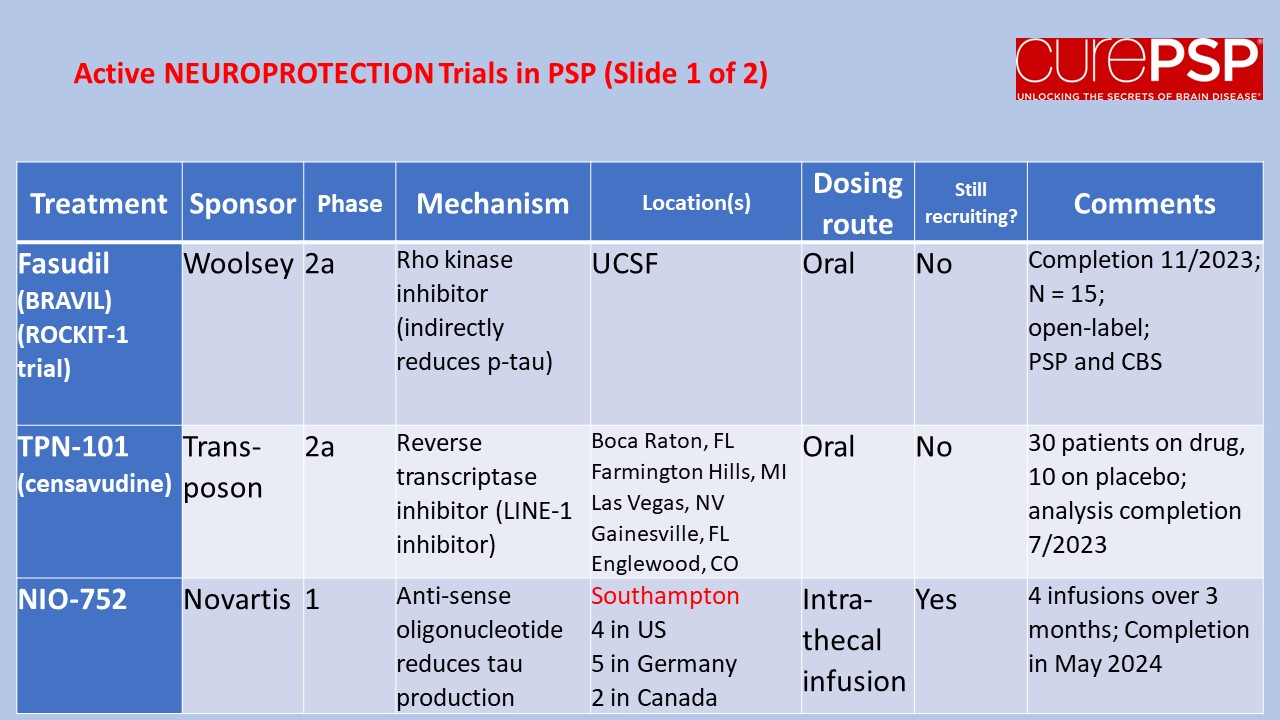
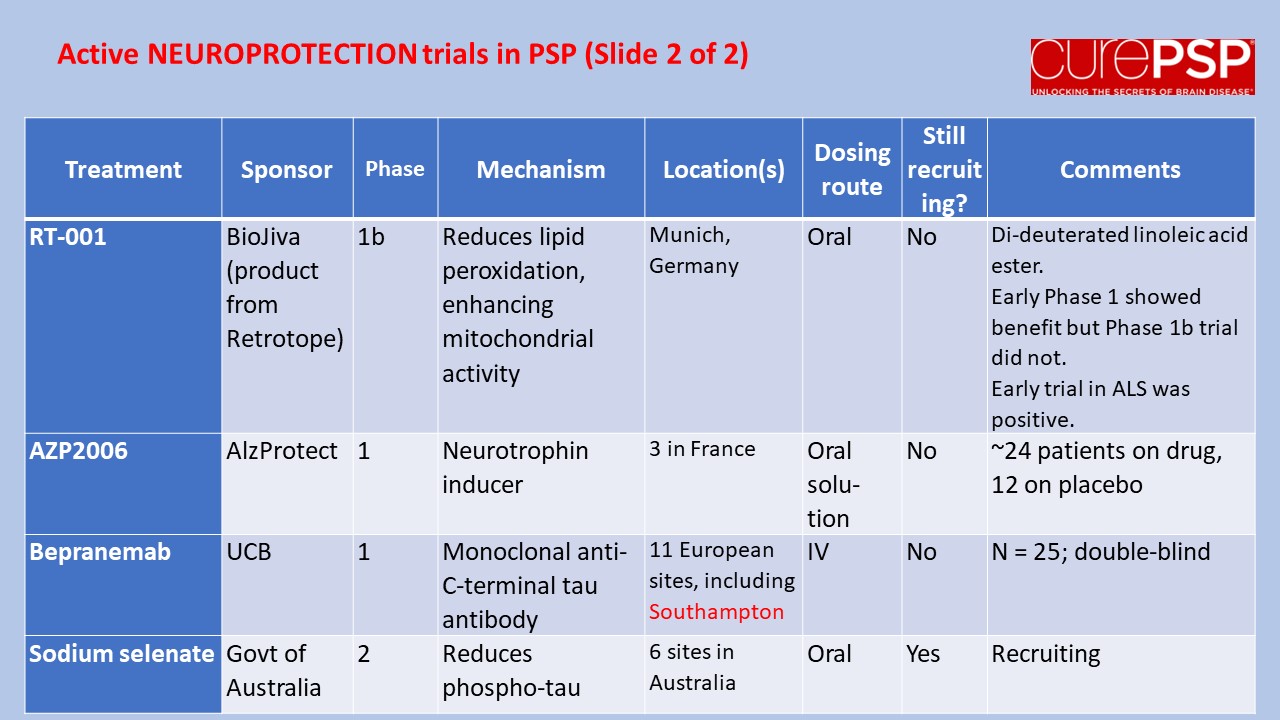
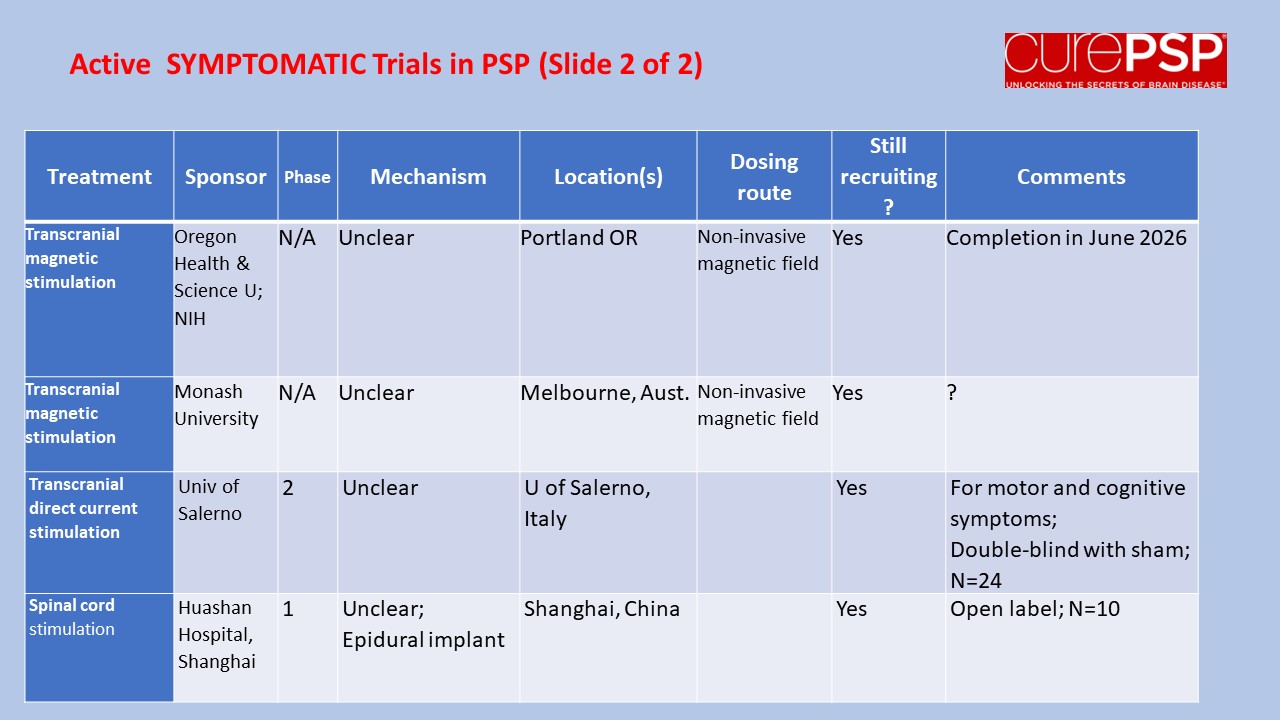
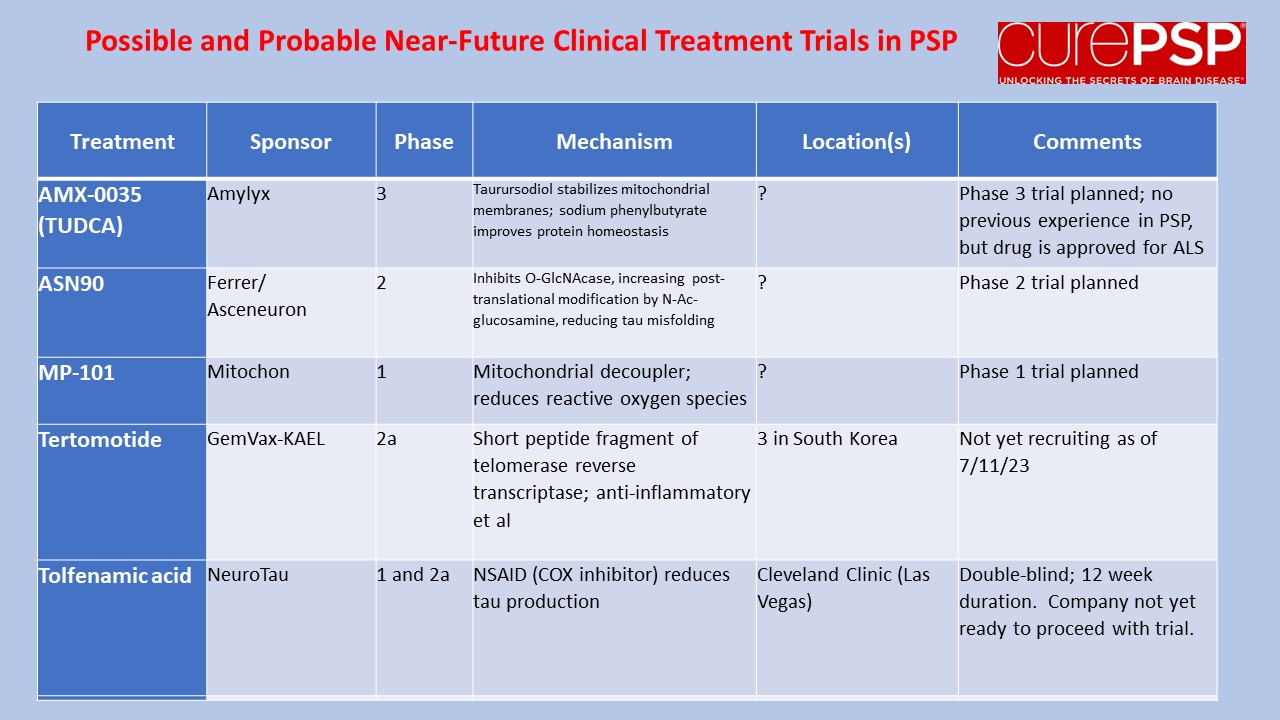
My last post was a nerdy list of pathogenetic mechanisms in PSP along with the statement that treatments to address most of those mechanisms are in the clinical pipeline. One of you wrote in to kick me out of my lofty, scientific detachment, asking just what those treatment candidates are. So here’s a list.
The first four panels are active trials and the last is future trials.
The first two and last panels show neuroprotection trials (i.e., to slow disease progression).
The third and fourth show symptomatic trials (i.e., to help the symptoms without affecting the underlying disease process).
For current information on how to enroll, visit clinicaltrials.gov and search on the drug and/or sponsor and/or “progressive supranuclear palsy.”
I watched a scientific presentation today in which the speaker started off by summarizing the leading theories of PSP’s pathogenesis. That means not the external influences such as the genes received from one’s parents or whatever toxins or other stresses might help cause PSP in susceptible people. Rather, it means the abnormal processes set in motion and operating inside in the brain cells leading to their dysfunction and eventually, their death.
Here’s a quick rundown for you:
Tau splicing. The tau protein is encoded by the MAPT gene, which has 14 sections called exons encoding separate fragments of the final protein. These protein fragments are then stitched together, but sometimes one or more of them is omitted by design. In healthy people, the product of exon 10 is included in about half of the final tau molecules, but in the tau tangles of PSP, that fragment is nearly always included. This makes the tau more likely to aggregate.
Tau post-translational modifications. Many or most proteins have very small molecules attached to them at specific points to regulate their function and direct their folding pattern. The abnormal tau of PSP has phosphate and other molecules in inappropriate places. This could help explain the abnormal folding, which in turn produces toxic aggregates.
Tau degradation. The normal “garbage disposal” systems of brain cells gets rid of proteins or organelles (the tiny structures in cells that perform specific functions) that are either overproduced, defective or just worn out. There are two basic kinds of such systems, the ubiquitin-proteasome system and the autophagy-lysosomal system. Neither works as well as it should in PSP. This allows abnormal tau and other toxic molecules to accumulate.
Intracellular tau spread. In many neurodegenerative diseases, the abnormally folded tau can travel from one brain cell to another, causing normal copies of those molecules to misfold in a similar fashion. This creates a kind of chain reaction spreading the damage widely. The misfolding pattern of the tau is specific to each of the tauopathies.
Mitochondrial dysfunction. The mitochondria are the organelles in the cells that harvest energy from sugars with the help of oxygen. In PSP, they function abnormally, possibly because of their own genetic mutations, possibly because their biochemistry is particularly sensitive to certain toxins in our environment. Mitochondrial dysfunction doesn’t just deprive the cell of energy – it also produces toxic compounds such as free radicals that damage other cell components.
Gene expression errors. The most recently discovered pathomechanism has to do with abnormal regulation of access of the cell’s protein-making machinery to the DNA “blueprint.” That process is normally regulated by proteins collectively called “chromatin,” which coat and intertwine with the DNA in the nucleus. One way the abnormality might work is that abnormal chromatin permits inappropriate access to certain genes that stimulate the immune system, producing a harmful inflammatory reaction in the brain.
All of these pathogenetic mechanisms except the first are currently being addressed by drugs in advanced stages of the development pipeline. I really don’t know which horse to put my money on.
CurePSP recently received an inquiry from a PSP caregiver who had evaluated the individual with PSP twice over a 6-month period using the PSP Rating Scale and needed some guidance in interpreting the results. We had to tell them that the PSPRS is designed for use only by neurologists experienced in evaluating people with movement disorders and eye movement disorders, so the scores they generated cannot be relied on. Besides, the second score was about 30 points (on the 100-point scale) worse than the first, and no one with PSP progresses that quickly. So, they must have administered the PSPRS incorrectly.
People with PSP and their caregivers who ask about the PSPRS are advised to pass it along to their neurologists, who can decide if they want to use it. The PSPRS is still the world-wide standard “outcome measure” for PSP treatment trials and the rating standard for observational research. But it has reached only limited acceptance in ordinary neurological practice outside of academic settings because it takes 10-15 minutes to apply, and most neurologists don’t have that kind of time in a visit to devote to it.
But don’t despair, you neurological do-it-yourself-ers. There’s a newer scale called the Cortico-Basal Functional Scale (CBFS), which is designed to be completed by the patient and caregiver and works just as well for PSP as for CBS. It’s not quite as precise as the PSPRS because it relies only on subjective symptoms and experiences, but it’s still quite reliable in assessing the severity of PSP and tracking its progression. It has more potential to be adopted by neurologists for routine care because it can be completed at home the day before the visit or even in the office waiting room. You can download it via the link above, complete the 31 multiple-choice questions and bring the completed form to your next neurology visit.
My last post, on current imaging techniques for PSP, was kind of technical and its last paragraph promised that the next one would be something softer. But someone just sent a comment asking why I didn’t mention dopamine transporter (DaT) imaging. So I’ll defer the low-tech post and in this one I’ll explain why I omitted DaT scan from my previous post. Here’s why: It’s not useful in distinguishing PSP from its likeliest diagnostic alternatives.
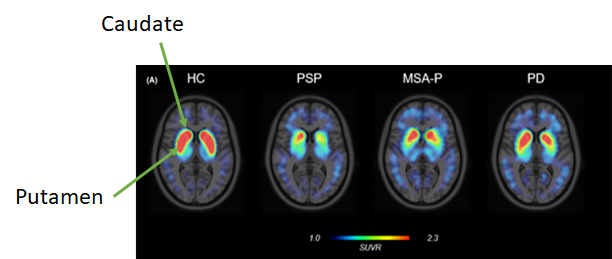
The dopamine transporters are molecules in the caudate and putamen (which together are called the striatum). See the highlighted structures in the image just below:
The brain cells bearing the DaT molecules have their cell bodies down in the substantia nigra of midbrain and send their axons up to the basal ganglia to synapse in the caudate and putamen, where they use dopamine as their neurotransmitter. In PSP, MSA, CBD, dementia with Lewy bodies and some others, those neurons are among the first to die and in Parkinson’s they’re not the first, but they’re the most important. So any imaging technique that reveals those neurons will be abnormal in all those diseases.
In the images below, the red and yellow areas represent the greatest presence of DaT molecules. The “head” of the comma-shaped thing is the caudate and the “tail” is the putamen. They’re nice and chunky in the leftmost image, labeled HC for healthy control. People with PSP, MSA-Parkinson type and Parkinson’s disease have lesser DaT signals, and the differences among those are just related to the severity of illness in those three individuals. They all have the same basic abnormality.
So, the DaT scan can quantify the severity of disease but cannot distinguish among the various neurodegenerative causes of Parkinsonism. Of course, for purposes of patient care, one can quantify PSP severity more easily, cheaply and usefully with just a history and exam.
What DaT can do, and this is its only official, FDA-approved use, is to distinguish essential tremor, where the dopamine-producing neurons are normal, from degenerative causes of tremor, where of course they’re not. Uses of the DaT scan that have not been approved by the FDA because of insufficient data are to distinguish degenerative Parkinsonism from normal-pressure hydrocephalus, drug-induced Parkinsonism, hypothyroidism and a bunch of other things that cause muscle rigidity and slow movement for reasons other than loss of the dopamine-producing neurons. But all of those things can be diagnosed in other ways.
Another difficulty with the technique is that many commonly-used neurological drugs such as antidepressants can cause false-positive DaT scans by blocking the dopamine transporter.
Bottom line: Other than to distinguish unusual cases of essential tremor from degenerative Parkinsonism, most movement disorders specialists rarely or never order DaT scans for routine patient care because they add nothing to a properly performed history, neurological exam and MRI.
A very active area of research right now is how various imaging techniques (“scans” in English) can and cannot assist in distinguishing the atypical Parkinsonian disorders from other conditions and from one another. Yes, this is important for clinical care and counseling. But even more important right now is that until we have specific treatments for these diseases, we need accurate diagnosis in living people. This is important for laboratory researchers who want to know the true diagnosis of the patient who supplied a fluid sample, and to the designers of clinical trials who want to make sure the patients in their trials have the disease for which the treatment was designed.
Dr. Jennifer L. Whitwell, a radiology researcher at the Mayo Clinic in Rochester, MN has just published a very helpful review of that topic in Current Opinion in Neurology. It gets pretty technical, but here are the takeaways with, of course, my own editorial contributions:
Measurement of atrophy by MRI:
The magnetic resonance Parkinsonism index (MRPI), especially its updated version, the MRPI 2.0, gives excellent differentiation of PSP from non-PSP, with an area under the receiver operating curve of 0.98. (That statistic is 1.00 for perfect accuracy and 0.50 for no better than a flip of a coin. For a bit more explanation of the AUROC and a graph, see this post from last year.)
Routine MRI can also be used to compare the various PSP variants with regard to atrophy of specific brain structures. Atrophy of the brainstem is worse in PSP-Richardson syndrome, PSP-CBS and PSP-frontal than in the others. This information could be useful in treatment trial design because some PSP variants progress faster than others. So, a trial of a disease-slowing treatment could potentially determine which patients have which variants and adjust the statistical analysis of the treatment outcomes accordingly.
MRI shows that atrophy in PSP-Richardson’s syndrome starts in the midbrain, (the upper part of the brainstem where the substantia nigra and the vertical gaze centers reside), followed by a succession of other areas of the brainstem, cerebellum and basal ganglia, before finally reaching the cerebral cortex. But two PSP subtypes involving relatively more cortical function (the frontal behavioral type and the corticobasal syndrome type) spread into the frontal cortex much earlier in the process, although like PSP-RS, they start in the midbrain. These observations could help guide designers of other imaging-based diagnostic tests for PSP.
Measurement of metabolism by fluorodeoxyglucose positron emission tomography (FDG PET): The pattern of reduced brain tissue metabolic activity in PSP can be distinguished from normal with an AUROC of 0.99. Distinguishing PSP from corticobasal syndrome, multiple system atrophy and Parkinson’s disease is more difficult but still useful at 0.90.
Measurement of tau deposition by PET: A PET technique that reveals the location of abnormal tau in Alzheimer’s is already in standard clinical use for AD, but it doesn’t work well for other tauopathies. Two techniques designed for PSP are expected to enter pivotal clinical trials in the next few months. In small studies, the two ligands called PI-2620 and APN-1607 (formerly called PM-PBB3) have shown good results in distinguishing PSP from the other tau disorders and Parkinsonian disorders. (A PET ligand is the chemical injected intravenously that sticks to the brain chemical of interest, allowing its location to be mapped.) But these two ligands can occasionally give conflicting results when given to the same patient, so the AUROCs from the upcoming trials are eagerly awaited.
Measurement of iron deposition by MRI: This technique, called quantitative susceptibility mapping (QSM), is not part of a routine MRI, but it can be performed with the same machine. Its PSP-diagnostic results are not quite as accurate of the ones above, with an AUROC of 0.83 for distinguishing PSP-Richardson’s syndrome from Parkinson’s disease. I assume the AUROCs for other kinds of PSP, especially PSP-Parkinsonism, are worse, but work continues on this technique.
Functional MRI: The spread through the brain of abnormal tau, and with it the damage of PSP, proceeds along “functional networks.” That means that after first affecting the substantia nigra in the midbrain, the damage proceeds to areas most closely yoked to it in terms of simultaneous electrical activity. Those physiological relationships have been delineated by “functional MRI” technique, where a person is given various mental or motor tasks to perform while in the MRI machine. The MRI software is set to measure not physical or chemical structure as for routine MRI, but blood flow, which correlates exquisitely with brain tissue electrical activity. These observations could potentially allow researchers to assess the effects of experimental drugs on the immediate or longer-term pattern of brain cell activation in people with PSP.
Exciting developments all, and I apologize for nerding out on you yet again. But as one of this blog’s commenters said a few years ago, “Thanks, Dr. Golbe, for respecting our intelligence.” Still, I’ll try softer stuff next time.
Yesterday’s post about training general neurologists to recognize PSP early in the course elicited a reader comment about one patient whose first symptom of PSP was dry eyes. The diagnosis of PSP remained elusive through multiple years and multiple doctors. So, a word about that symptom seems in order.
Dry eyes are common in the general population, usually as a result of insufficient tear production, but in PSP the problem is too little blinking – about 20 percent of the normal frequency for age. This causes inflammation of the conjunctiva (the very thin, transparent layer covering the cornea, the sclera and the inside of the eyelid) and a sensation of dryness or itchiness. Part of the eye’s reaction is to increase the production of the watery component of tears, but that doesn’t help the loss of the oily component, which comes from a different set of glands.
Often, the eyes’ reactions also include a reflexive increase in blinking when awake. This is helpful, but during sleep the lids may remain open because of the basic eyelid dysfunction of PSP. The resulting drying during the night creates more inflammation than can resolve over a waking day’s-worth of increased blinking.
An ophthalmologist or optometrist evaluating someone for dry eyes may fail to evaluate the blink rate. Or, if the person has reflexively compensated by increasing the blink rate, the doctor may interpret that as only the normal reaction to inflammation of the conjunctiva.
So, how can an eye doctor start to suspect early-stage PSP in someone with dry eyes? Examine the movements of the eyes themselves! The earliest ocular motor abnormalities of PSP (and here I’m mainly referring to PSP-Richardson’s syndrome, which comprises about half of all PSP) are slow saccades, the “round the houses sign” and square-wave jerks. None of these require special equipment – just a little book knowledge, a few teaching videos and some practice. Actual limitation of downward, voluntary eye movement – the “palsy” in the name — arrives a year or two later. Here’s what those three early signs look like:
Slow saccades: A saccade is simply an eye movement, and in PSP they’re slow enough for the doctor to see them in progress, while a normal saccade is too fast – the doctor only sees the starting and ending positions. In PSP, the slowness is first and worst in the downward direction.
The “round the houses sign” is where an attempt to move the eyes downward takes a curved path, as if trying to avoid a direct downward movement. Probably a more sensitive way to elicit this sign is to ask the patient to perform a diagonal saccade, say from upper right to lower left. The eyes will first move mostly horizontally and then, once the patient’s visual system realizes that the target isn’t being reached, will force the eyes to move downward to compensate.
Square wave jerks are pairs of short, involuntary horizontal eye movements performed by both eyes together up to 20 times per minute. The eyes first go to the right or left (seemingly randomly) and after a very brief pause, return to the starting point. They’re especially pronounced when staring at an illuminated target in an otherwise dark room. They occur in most people with PSP but also in a minority of those with other Parkinsonian disorders and even in a few healthy elderly people.
So, an ophthalmologist or optometrist seeing a patient with dry eyes should look for these things and if even one of them is present, refer the patient to a neurologist (preferably a movement disorders sub-specialist) to investigate further for PSP.
I don’t need to remind this readership of the advantages of earlier diagnosis of PSP, even if a cure isn’t yet at hand, but I’ll do so anyway: Access to disease-specific counseling and information; ability to plan one’s financial and living arrangements; avoidance of useless, risky and expensive diagnostic tests and treatments; access to PSP clinical trial enrollment; and the intangible benefit of just knowing what you’re dealing with.
Now that I’ve shared the details of a work week and my breakfast menu, I’ll share my innermost thoughts. Or maybe not my innermost — just some neurology-related thoughts that have been occupying a lot of my conscious moments lately. No, these thoughts aren’t about anything very interesting, unless you have an atypical Parkinsonian disorder (APD), want the best possible care and want it fast.
Here’s the background: As CurePSP’s Chief Clinical Officer, I serve on the Steering Committee of CurePSP’s Centers of Care (CoC) network. That’s a group of 28 movement disorders referral centers in the US and 2 in Canada with particular interest and qualification in the care of PSP, CBD and in many cases, MSA. (The centers are already in place – CurePSP does not create or operate them, but it does provide each CoC a token $5,000 per year to help defray expenses.) The network’s mission is to improve the quality of, and access to, first-rate care for these diseases, both at our own centers and generally. In 2021, the group published a “best practices” document on the symptomatic management of PSP and CBD. (That means how to treat the symptoms to make patients’ lives better until we actually have a cure.)
One of the current goals of the CoCs is to make it easier for people to be evaluated by a doctor with the training and experience needed to tell them if they have PSP, CBD or MSA, and if not, what they do have, and then to advise them on prognosis and management. One way the CoCs try to do that is to expand our ranks until we have a center within a reasonable drive of most of the population. Another is to reduce the wait for appointments, which among the CoCs averages 3½ months and for 14% of the CoCs, exceeds 6 months.
Why is 3½ months (or 6!) too long? Because PSP, CBD and MSA can progress quickly. The average patient with these diseases survives only about 3-4 years after receiving the correct diagnosis, so 3½ months is a big chunk of that. Could an oncologist tell a woman with an abnormal mammogram to wait 3½ months to be seen? No, and they don’t. Even (or especially) the busiest cancer specialists have figured out how to see new patients within a few days.
We would like the wait for an initial appointment for suspected APDs not to exceed one month. One of the ideas the CoCs have been batting ideas around in our Zoom calls is to reserve a couple of hours each week just for patients with known or suspected APDs and then, to prevent those time slots from being overwhelmed, to reduce demand. In other words, provide referring physicians – usually general neurologists – with a convenient diagnostic algorithm for the APDs to allow them to chose the next diagnostic tests themselves and initiate management. Then, a confirmatory evaluation with the subspecialist can wait 3½ months without much risk of harm.
To that end, the CoCs are working on a practical guide for general neurologists on how to diagnose all the APDs, and I do mean all – not just to recognize PSP, CBD and MSA, but also to recognize 49 other progressive disorders in adults that can mimic aspects of PSP, CBD and MSA.
It’s hard to get busy physicians to sit down and read a textbook, especially about diseases they’re not going to see very often. So, we’re describing a step-by-step process in the form of a few charts to allow general neurologists to apply a diagnostic decision tree in real time to 55 possible disorders and initiate treatment. We’re deep into the process as a group, so I can’t say more right now, but I will as soon as I can.
What I can say now is that the final product will be available for free on the CurePSP website and by a link from this blog.
Tertomotide, or GV-1001, is a small polypeptide, which means that it’s a string of amino acids, like a protein. But with only 16 amino acids, it’s much smaller than any protein. In fact, it’s a critical fragment of a protein, in this case an enzyme called telomerase reverse transcriptase. The drug was originally developed as an anti-cancer drug. Initial results there were unfavorable, but trials continue. In the lab, tertomotide protects brain cells growing in a dish from various insults including free radicals and inflammatory attack.
As many of the effects of tertomotide might be relevant to neurodegenerative diseases, it has also been tested in Alzheimer’s disease. A study published in 2021 found the drug to slow the rate of decline by upwards of two-thirds on a standard, 40-item test for AD called the “Severe Impairment Battery.” In one patient sub-group, the rate was slowed from 3.7 points to 0.1 point, or 97%.
Normally, a drug trial attempting to slow the rate of progression of a neurodegenerative disease would be happy with a 25 or 30% slowing and overjoyed to see 40%. So, this extravagantly positive tertomotide result in AD might be too good to be true, and in fact none of the other five tests of various aspects of AD gave a favorable result to any statistically significant degree. So, more study is needed. At least there were no side effects among the 55 patients receiving the active drug.
These results have encouraged not only further trials in AD, but also a trial in PSP! It’s not yet recruiting and it’s taking place only in South Korea. (Sorry — but glad for the South Koreans.) The study will randomly assign 75 patients to high dose, low dose and placebo groups. As in the AD trial, the drug is injected subcutaneously once a week for 4 weeks and then every 2 weeks for a total of about 6 months. The outcome will be measured by the rate of progression on the PSP Rating Scale.
The drug company’s website is here. http://www.gemvax.com/bio_en We wish them well. I’ll report back when I know more.
The brain bank for neurodegenerative disorders at Mayo Clinic in Jacksonville, Florida houses the world’s largest collection of brains from people with PSP and with CBD. It currently makes available to researchers world-wide about 9,000 brains overall, including about 3,000 with Alzheimer’s disease, 2,000 with Parkinson’s disease or Lewy body dementia, and nearly 3,000 with the neurofibrillary tangle disorders listed below. The table also shows the number of each along with some random comments.
First, an FYI: Don’t assume that the relative proportions of these disorders in the brain bank reflect their relative prevalence in the population. Brain banks disproportionately attract unusual conditions where the diagnosis during life was unclear and the family is seeking an accurate, expert final diagnosis as much as they’re making a charitable gift to research.
PSP
1,762
CurePSP encourages brain donations to this brain bank and subsidizes the cost of brain removal and shipping for families who cannot afford those costs.
Corticobasal degeneration
358
It can be very difficult to distinguish CBD from PSP during life. About half of people with corticobasal syndrome during life turn out to have CBD at autopsy.
Neurofibrillary tangle dementia
267
This is an umbrella term for dementing illnesses with NFTs other than Alzheimer’s disease.
Frontotemporal dementia with a mutation in the MAPT (tau) gene
86
A rare, hereditary form of frontotemporal dementia. Depending on the type and location of the mutation within the MAPT gene, it can closely mimic PSP both during life and at autopsy.
Chronic traumatic encephalopathy
78
Caused by repeated blows to the head in susceptible individuals. The location of its neurofibrillary tangles is very different from that of any of these others or Alzheimer’s disease.
Pick’s disease
77
This is a non-hereditary form of frontotemporal dementia, with more motor and language deficits than most other forms of FTD.
Globular glial tauopathy
46
During life, this produces speech apraxia, parkinsonism, behavioral changes, eye movement changes. It also has lower motor neuron changes similar to those in ALS.
Total:
2,674
These are impressive numbers and you might ask why CurePSP is still encouraging families to donate their loved one’s brain. In other words, why do we need more? It comes down to statistics and genetics. Stay with me here and — spoiler alert — some details in the next paragraph may be unpleasant to think about:
Each donated brain is sliced into right and left halves and then into one-centimeter-thick slabs. One half is put into a preservative solution for two weeks before it’s firm enough to be sampled for examination under a microscope. That establishes the diagnosis, and the pathologist at the brain bank sends a detailed report to the family and local physician if the family so requests. The other half is frozen for use in research on the chemical components, including proteins, DNA and RNA.
About 15 years ago, CurePSP funded a project using DNA extracted from the brain samples then available from the Mayo brain bank along with some from other, much smaller brain banks. The research group created for that was called the PSP Genetics Consortium. Its 2011 publication reported that variants in five genes were more frequent in PSP than in controls without PSP. One of those variants confirmed the previously-discovered association of the “H1 haplotype” with PSP.
(I’ll digress for a bit here. The H1 haplotype is a span of chromosome 17 that encompassing the MAPT gene and about a dozen others. The variantisn’t a simple substitution of one nucleotide (as each “letter” in the genetic code is called) for another. Rather, it’s an “inversion,” a long string of genes occurring in reverse order on its chromosome. The H1 haplotype actually is present on both copies of chromosome 17 in about 60% of European-derived populations but in 88% of those with PSP — a statistically significant difference but far from a full explanation. End of digression.)
That 2011 analysis found another variant at a specific locus inside the MAPT gene plus three others on different chromosomes, each of which performs functions consistent with what’s known about how PSP works. They’re called EIF2AK3, MOBP and STX6.
Two other genes called SLCO1A2 and DUSP10 have since been found to increase PSP risk and one other, TRIM11, has been found to affect onset age of PSP, but not the chance of developing the disease in the first place. And that’s it so far, other than a bunch of mutations in MAPT associated with very rare, strongly-inherited, familial forms of frontotemporal dementia closely mimicking PSP.
The conundrum is that even adding the influence of all of these known gene variants together can’t explain the magnitude of population’s prevalence of PSP, small though it is. One possibility is some strong, unidentified environmental factor. Another is that many more PSP-risk-conferring genes, each with only a tiny statistical effect, await discovery. They wouldn’t have appeared in the 2011 analysis for lack of enough brain samples to reveal their weak statistical “signals.” That analysis relied on comparing the frequencies of gene variants between the brains from people with PSP to a group without PSP. Any variant with a difference greater than what might be expected by chance is considered a genetic “hit.”
The Genetics Consortium tried solving that problem by doing whole-exome and whole-genome sequencing in the original set of brain samples plus a few hundred others that had accrued since. Those studies are not yet published, but my information is that they have produced at most one more hit, called TREM2, which was already known to be a risk gene for Alzheimer’s disease.
So, the solution is more samples from more donated brains with PSP. Barring some breakthrough in genetic technology, that’s the only way we’ll ever have enough samples to compare with controls to tease out more hits — gene variants each contributing only a tiny degree of risk.
Why bother, you say? The main reason is that identifying a risk gene may point to a specific protein or biochemical pathway (a set of closely related chemical reactions in the cell) that, when impaired for any reason, results in the disease. Then, researchers have a “drug target” on which to focus their search for ways to improve the performance of that chemical or pathway.
Another reason to bother with the genetics of PSP is that if we identify enough risk genes, we can create a diagnostic test panel based on a “polygenic risk score.” That’s where DNA from someone suspected of having PSP would be tested for all of the gene variants known to contribute PSP risk. If enough of them are present (or present in a specified combination), the diagnosis is made* and the person can enter a clinical trial at an early stage or can receive a disease-slowing drug that might be available by then.
So, if you have PSP, I hope we find a cure during your lifetime. But if we don’t, please consider making (non-binding) arrangements to donate your brain to a research-oriented brain bank like the one at the Mayo Clinic in Jacksonville. More information is available here.
* It might seem that having a set of genetic variants associated with PSP would be pretty good proof of the diagnosis. But it turns out that a majority of people with mild brain cell damage of PSP at autopsy never actually had outward signs of PSP, even into their 80s. During life, such a person would have a positive genetic diagnosis but their outward neurological symptoms might be caused by something else. More on this in a future post.