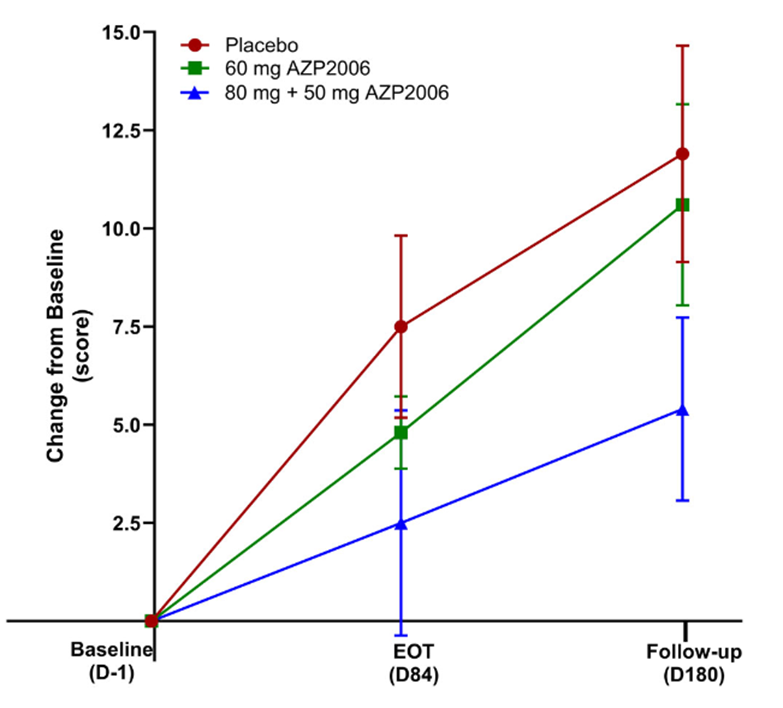
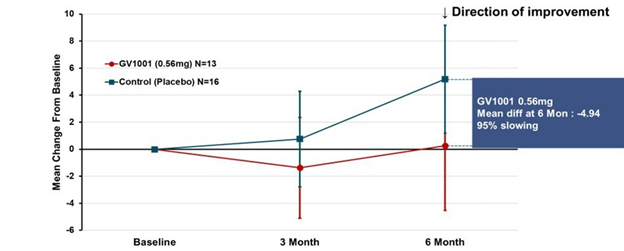
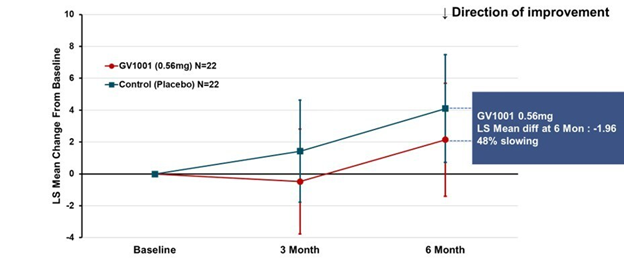
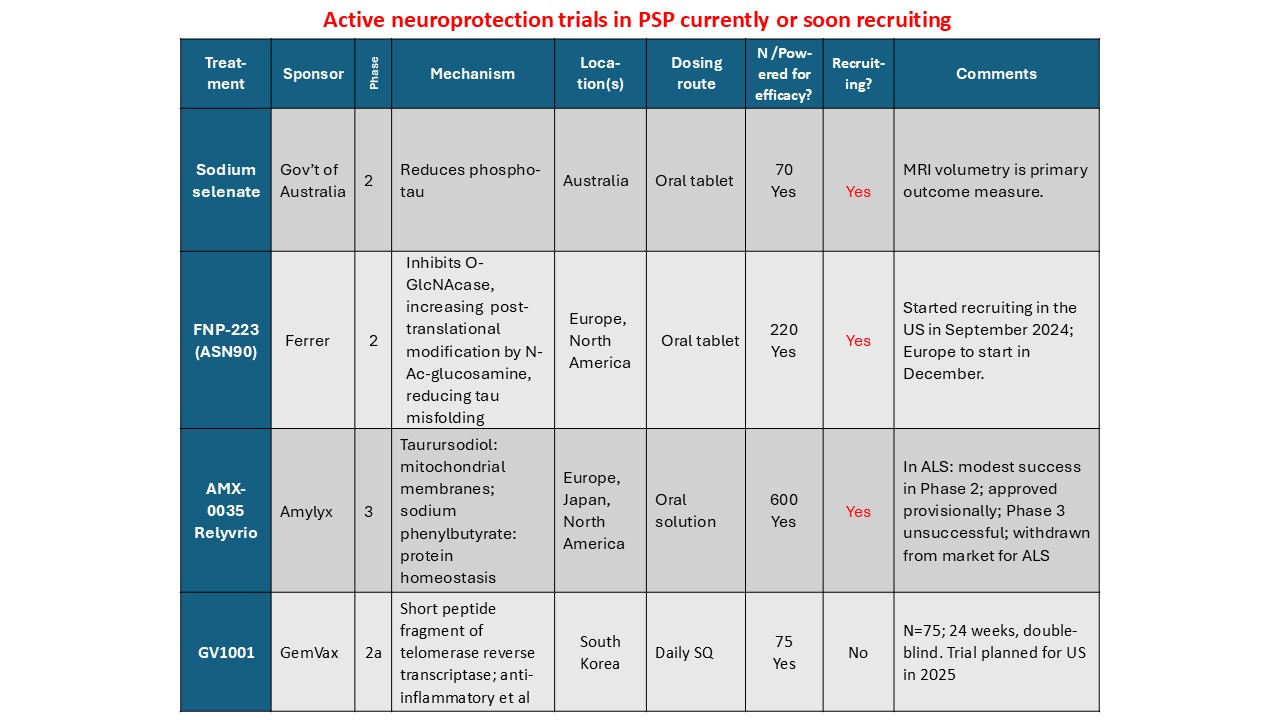
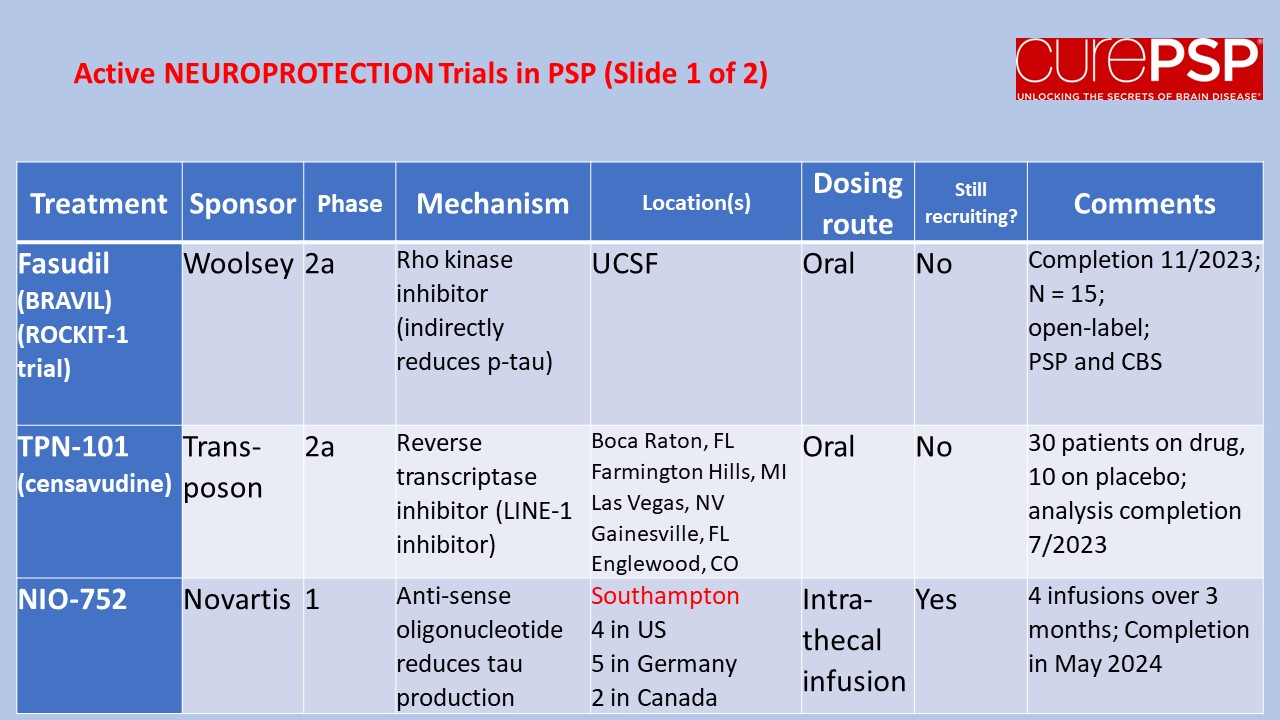
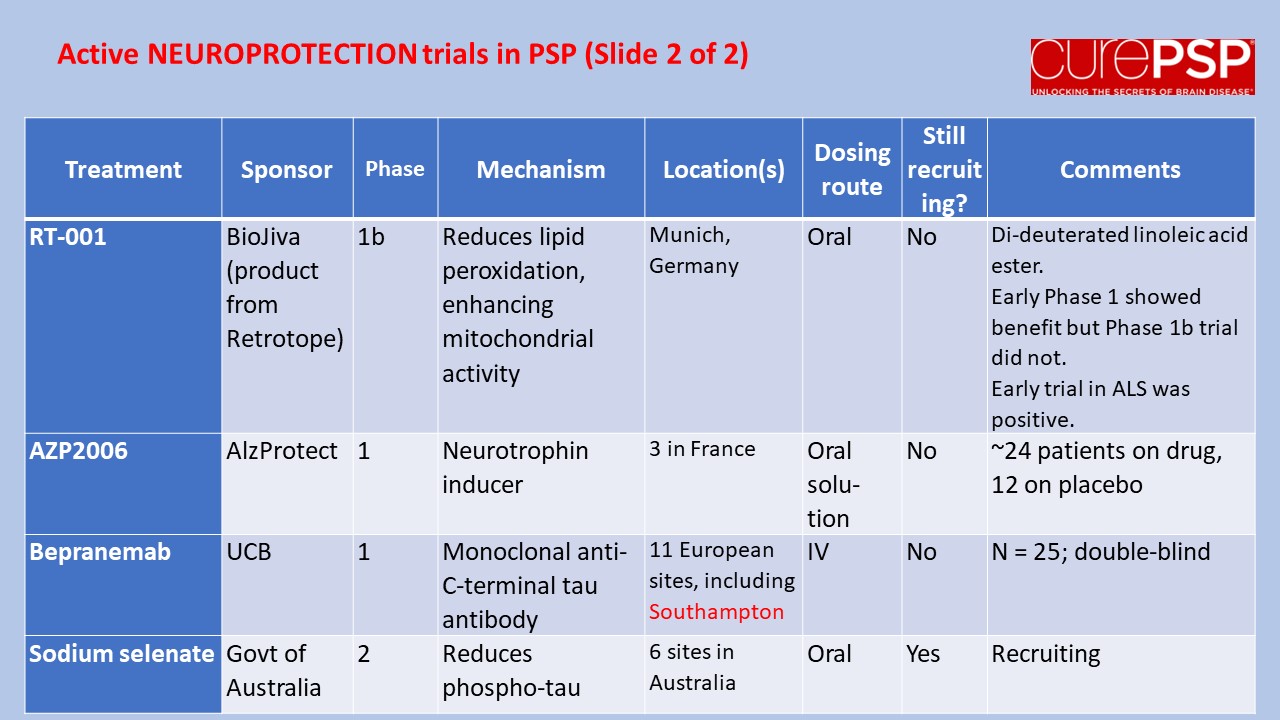
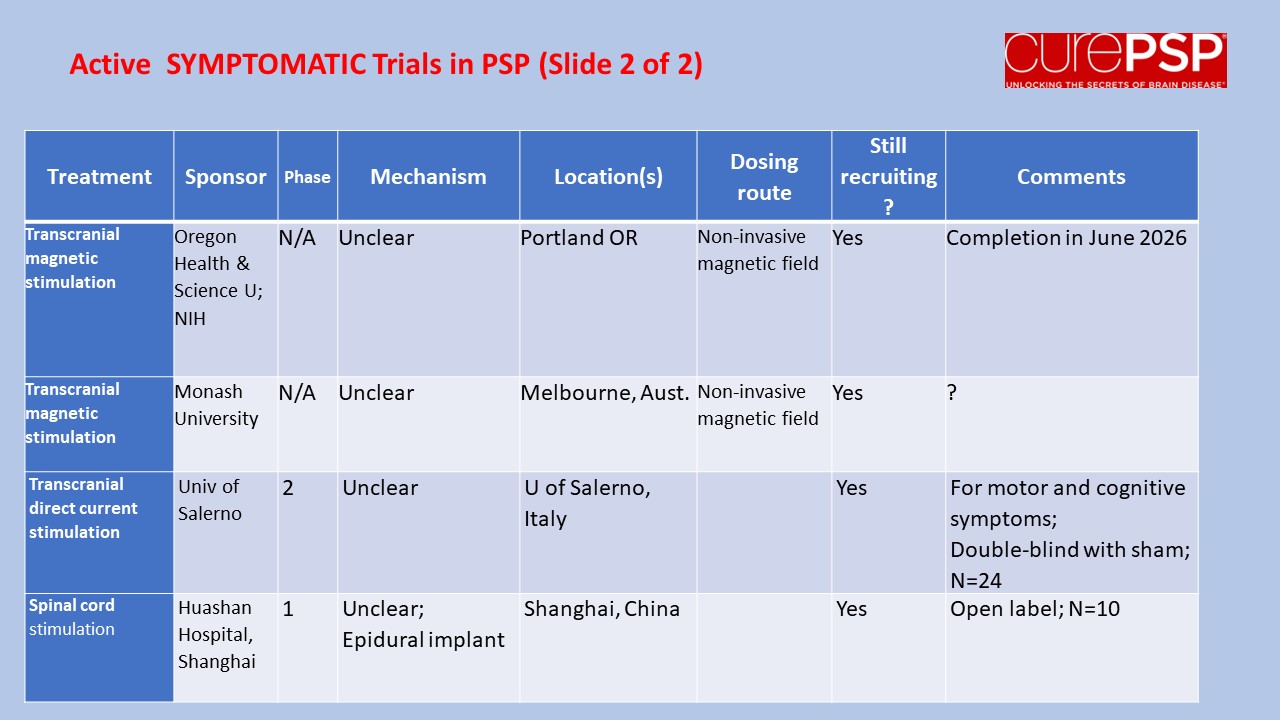
Prompted by a reader’s comment a few days ago, I thought I should write about the difference between “neuroprotective” and “symptomatic” treatment. The distinction is relevant to the PSP treatment trials about to start and to one’s decisions about whether to volunteer for them.
Neurodegenerative diseases, by definition, progress over time and neuroprotective treatments attempt to slow that process, not to provide relief from existing symptoms. In theory, such treatments could be so miraculously effective as to halt the process in its tracks, but a realistic best-case target given our current understanding of these diseases is a 40%-50% slowing of the rate of future progression. Most PSP trials are designed to detect about a 25%-30% slowing. A trial large enough to detect more subtle degrees of slowing would be prohibitively expensive.
For a drug to improve the existing PSP symptoms or disabilities, as levodopa improves those of Parkinson’s, for example, would require replacing a molecule deficient in the brain cells that still survive and function, or stimulating the surviving cells to work harder, or modulating the activity of other, healthy, brain cells to partly compensate for the effects of the damaged cells. Such drugs do exist for PSP, but their benefits are modest and temporary, and the underlying neurodegenerative process continues. In Parkinson’s, levodopa gives dramatic and long-lasting symptomatic benefit, but even there, the degenerative process continues unabated.
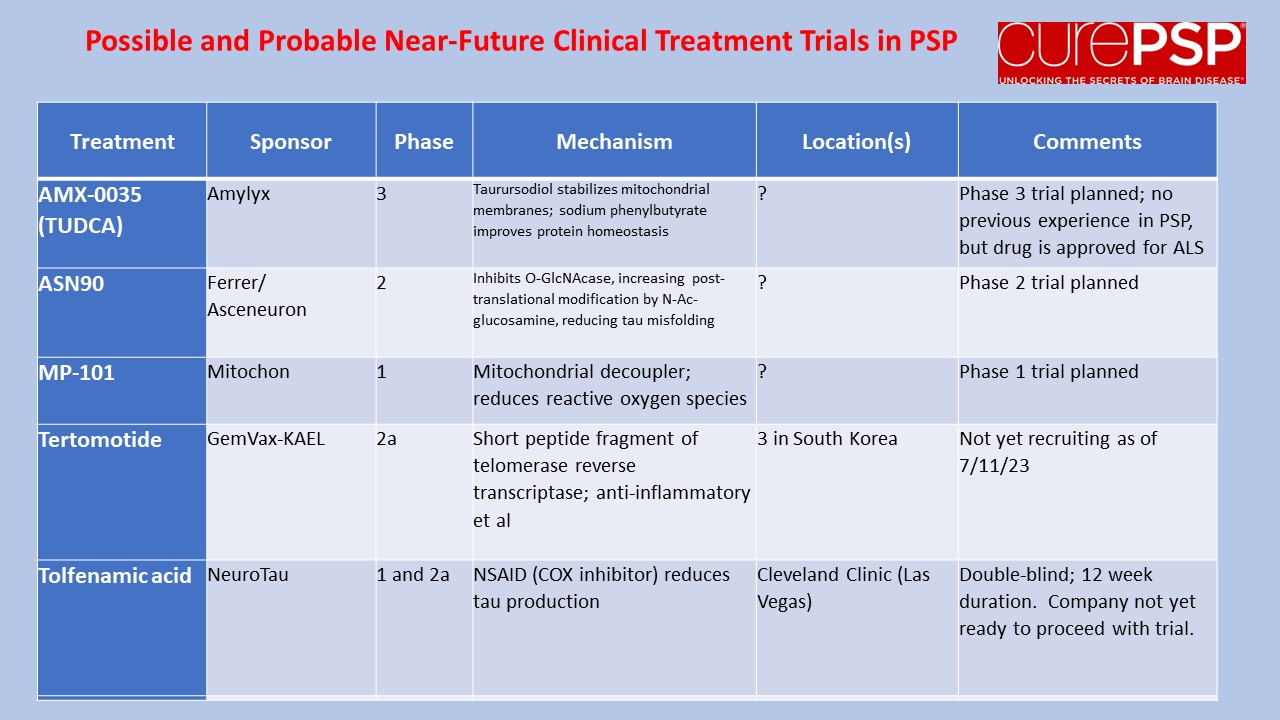
The graph below illustrates all this:
- The vertical axis is the PSP Rating Scale, where 100 is the worse possible score and the average rate of worsening is about 11 points per year. At a score of about 80, fatal complications such as pneumonia or severe urinary tract infections become very common. Note that to avoid displaying blank space, the axis starts at 30 points, not zero.
- The horizontal axis is years since the start of the treatment (the “baseline”). You can see that for purposes of this illustration, I’ve chosen a baseline PSPRS score of 40, which conforms to experience with previous trials and to the observation that the average person with PSP doesn’t receive that diagnosis until about three years after symptom onset.
- The blue line represents the course of the disease untreated. In a drug trial, there’s usually a placebo effect, but to keep things simple, the graph ignores that. Besides, that effect would dissipate over a couple of months at most.
- The orange line represents the course 30% slower than that of the placebo group. Note its shallower slope. Again, for simplicity I show the effect as starting immediately upon receiving the drug even though some neuroprotective effects may take a few months to get going.
- The green line shows a symptomatic effect, which in this example starts immediately and lasts years. I’ve semi-arbitrarily chosen its magnitude to be five PSPRS points and the time to maximum benefit as six months. At that point the rate of progression of the underlying disease continues unabated, but the five-point symptomatic benefit persists. Note that the participants on such a drug are doing better than those on the successful neuroprotective drug until a bit after the three-year point, when the lines cross, and the advantage of the neuroprotection continues to widen.
HYPOTHETICAL COMPARISON OF EFFECTS OF PLACEBO,
NEUROPROTECTIVE AND SYMPTOMATIC TREATMENTS OF PSP
I’ll emphasize that while the 11 points per year rate of progression is based on real data, the 30% slowing of the rate of progression is only an illustrative example for the purpose of this instructional exercise. The five-point symptomatic improvement is analogous to the magnitude of improvement of Alzheimer’s disease treated with cholinesterase inhibitors such as donepezil, galantamine and rivastigmine.
The death of a brain cell isn’t like an incandescent light bulb suddenly burning out – it’s more like a slowly fading LED bulb. During that “ill” phase, it might be possible for a candidate neuroprotective treatment to instead (or in addition) have a symptomatic effect.
With all that as background, here are some conclusions:
As you’d imagine, it could be difficult to tell neuroprotective from symptomatic (or placebo) effects, as they’re both being measured by the same PSP Rating Scale. But clinical trials in PSP try to anticipate this by testing for more objective evidence of slowing of brain cell loss, for example by assessing atrophy on MRI or spinal fluid levels of a protein called neurofilament light chain (NfL), which increases steadily in PSP and some other disorders. Placebo and symptomatic improvement would not be reflected in those diagnostic markers.
Neuroprotection trials also perform their first repeat exams in the first few weeks and months to look for a rapidly-appearing difference in PSPRS scores between the active drug group and the placebo group. (MRI and NfL would not be useful so soon after baseline.) However, it would be difficult to decide whether such a PSPRS improvement is placebo effect or symptomatic effect.
There’s another way to distinguish a placebo effect from a physiologic effect (we avoid the term “real” because placebo effects are also real in their own way). That’s to assess the participants for worsening couple of months after the trial’s end, when any symptomatic effect would have dissipated. I haven’t seen the PRESERVE (Novartis’ NIO752 trial) protocol and its clinicaltrials.gov entry doesn’t address the matter, but I expect that it plans to do this, if only to monitor any adverse effects of the drug.
Bottom line: In a Phase 3 trial in a neurodegenerative disease, separating true neuroprotection from symptomatic and placebo effects is tricky. In future blog posts, I’ll try to sort out that tangle for you if I can.
—
PS #1: For an excellent, very recent review of the placebo effect, see this paper, which is written in language easily comprehensible to educated laypersons.
PS #2: Disclosure: I consulted for Novartis from 2018 to 2020, but not since. I have never held stock or any other financial interest in the company. But I do hold a major emotional interest in seeing their drug work, so there’s that.