My post on June 24, 2025 was entitled, “I like Parkinson-like.” It was about a paper that was about to hit the stands (a Boomerism) authored by myself and Dr. Junaid Siddiqui of Cleveland Clinic. It proposed replacing the terms “atypical Parkinsonism,” “atypical Parkinsonian disorder” and “Parkinson-plus disorder” with a new term, “Parkinson-like disorder” (or “Parkinson’s disease-like disorder” because let’s not get TOO prescriptive).
The June blog post promised to make a copy of the paper available to you all once it was published. Better late (by seven months) than never. My mind was jogged by one of my current projects, preparing a lecture on the atypical Parkinsonian disorders in general that I will deliver at the University of Vermont in April. Also jogging my mind was that it’s been nearly two weeks since my last post.
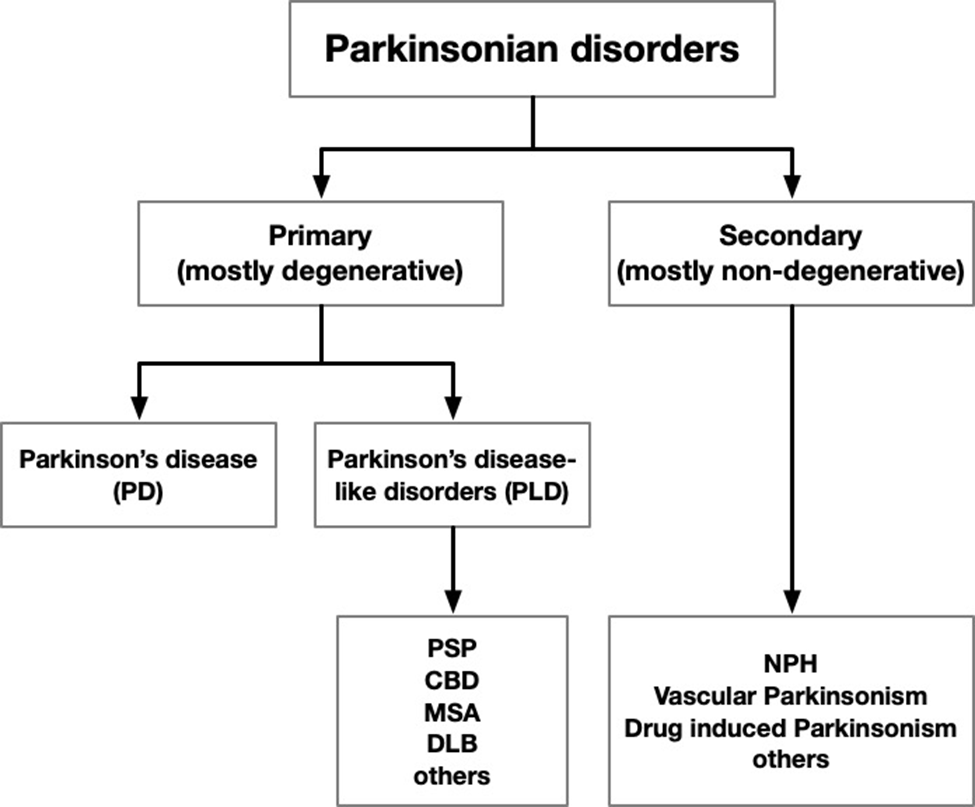
The pdf is at the bottom. If you have difficulty downloading or viewing it, here’s a link to the abstract and highlights and here’s a chart showing the gist of our proposed new terminology:
Yesterday’s post was about how the 10 most-frequently-visited news items in Parkinson’s News Today (PNT) of 2025 might relate to PSP. The list appeared here.
#5: FDA clears early trial of stem cell therapy for Parkinson’s
The treatment, called XS-411, is made from stem cells derived from multiple healthy donors and converted into dopamine-producing cells. Each patient would receive one injection of such cells into each side of the putamen, the brain area where the dopamine synapses are located. The FDA approved a Phase I trial in April 2025, but now, nine months later, clinicaltrials.gov lists two small studies in China but none in the US. Most current stem cell trials in neurology derive the cells from the patient’s own bone marrow or skin biopsy, so I really don’t know why the company, XellSmart Bop-Pharmaceutical Company of Suzhow, China, is using cells from people other than the patients themselves. Maybe they have a new way to suppress the immunologic rejection or maybe production using multiple donors is more easily scalable to serve large numbers of patients. Could XS-411 work in PSP? Perhaps it could help the same fraction who respond to levodopa, which is only a minority, and their benefit is usually modest and transient. The problem is that in PSP, unlike in PD, the cells in the putamen receiving the dopamine-encoded signals are degenerating along with the dopamine-producing cells. So, we need more research into injecting stem cells producing neuroprotective molecules or non-dopamine neurotransmitters.
#4: Program offers psychedelics as treatment for Parkinson’s
Ibogaine is a psychedelic drug legal only in a few states, and even then only for FDA-approved research use. Ambio is a company offering the drug to people with a wide variety of conditions on a “research” basis at clinics in Mexico and Malta. There’s no such listing in clinicaltrials.gov, so I can’t be sure of the protocol except that patients are charged $6,050 for a four-day treatment initiation at one of their clinics and “micro-doses” for the next six months. Based on the little information available, this fits the profile of many “research trials” of alternative treatments: a hefty fee, minimal pre-treatment evaluation or followup, no control group and no peer-reviewed publication. In this case, I must also wonder about the risk of habituation to the treatment itself, and where do you suppose you could buy something to satisfy that? Enough – you now know what I think about this drug for PSP, even if the initial $6,050 and the travel expenses are not an issue for you. To be sure, some alternative treatments do have legitimate potential, but when there’s a major risk of financial or medical harm (both of which apply here), their use should be confined to formal FDA-approved research protocols.
#3: Bacteria in digestive tract tied to cognitive decline
Parkinson’s disease is a natural candidate for causation by intestinal bacteria because the first stage of the disease, aggregates of the alpha-synuclein protein in neurons, starts not in the brain, but in the intestines and lungs. But PSP does appear to start in the brain. There’s been little research on gut bacteria and PSP, but something important was reported in 2023 from researchers in China and summarized in my 4/2/23 post. That short-term study found that replacing the colonic bacteria produced about a 10 percent benefit in the PSP Rating Scale score. The trial was too short and small to conclude anything about long-term slowing of progression. Bottom line: Although PSP does not start in the GI tract as PD does, gut bacteria may play a role and should be studied further for any therapeutic implications.
#2: Study identifies potential way to treat Parkinson’s constipation
Ghrelin is a string of 28 amino acids with many basic gastrointestinal functions including stimulating appetite at the level of the brain and defecation at the level of the spinal cord. The article reported by PNT teased out an important detail that could hold implications for treatment of constipation in those with PD. We don’t know if applies as well to the constipation of PSP, but I can say that some of PSP’s constipation is caused by degeneration of a cluster of cells in the lower spinal cord that are not involved in PD. One research study, from 2013, did find a role of ghrelin in multiple system atrophy but not in PSP or corticobasal syndrome. So, those few strands of evidence suggest that people with PSP will have to rely on traditional methods of keeping things moving – exercise, fluids, fiber, a stool softener, and avoidance of drugs that block acetylcholine synapses (“anticholinergics”). Many drugs in the last category are used for other PSP symptoms such as imbalance, vertigo, urinary incontinence and depression, so getting off those is a good topic for discussion at the neurologist’s office.
#1: Research shows disrupted mitochondrial DNA tied to inflammation
For decades, we’ve known that in both PD and PSP, the cells’ mitochondria malfunction and there’s excessive inflammation in the brain. But we don’t know which is cause and which is effect, or if they’re both effects of a common cause. The new study used a novel genetic technique to find evidence that it’s the inflammation causing the mitochondrial loss. In theory, the same study could be performed in PSP. A similar result would suggest that to slow PSP progression, targeting excessive inflammation might do better than those targeting mitochondria directly. Early in my career, I had narrowed my subspecialty choices down to movement disorders and neuroimmunology/multiple sclerosis. I chose the former because I was flummoxed by the complexity of the immune system. Little did I know . . .
A couple of weeks ago I posted my curated list of the top 10 PSP-related research developments of 2025. They appeared in two installments on 12/31/25 (#1-5)
Clearly inspired by my effort, the publication “Parkinson’s News Today” (PNT) has done something similar for Parkinson’s disease, summarizing their 10 most-often-visited (not necessarily the most important) news stories of 2025. Their article is very well supplemented with links to both basic explanations and to the PNT articles themselves. Those PNT items link, in turn, to original journal articles and to press releases from drug companies or research institutions.
PNT is owned by a for-profit medical communications company and its on-line publication is monetized by advertising, mostly from drug companies. So be aware that their choice of what’s important may be biased in favor of drug companies’ products and scientific narratives. No such concrete instances have hit me over the head, but the potential for a conflict of interest is there.
Armed with that caveat emptor, you can learn a lot from this list, which is numbered from least to most important. Here are my annotations for each item, pointing out the relevance, or lack thereof, for PSP:
#10: Virus long thought harmless may trigger Parkinson’s
The brain tissue in PSP looks very similar to that of “post-encephalitic Parkinsonism” (PEP), which was quite common from the 1920s to 1950s. Both feature tau-based neurofibrillary tangles and attack the basal ganglia, but PEP is static over decades. It appears to be a immunologically-based , residual effect of a brain infection called “von Economo’s encephalitis” or “encephalitis lethargica.” The virus itself has never been isolated or identified. The disease overlapped, but is different from, the great flu pandemic following World War I. Otherwise, despite ample search, there’s little to no evidence that any virus can cause PSP.
#9: Scientists develop weekly injectable implant for Parkinson’s treatment
The implant (actually a viscous, intra-muscular, self-injected liquid) provides levodopa/carbidopa (LD/CD) for a week. Unfortunately, most people with PSP respond little or not at all to levodopa/carbidopa. However, in those with the PSP-Parkinsonism subtype and few with other subtypes, there can be a useful response for a few months of daily dosing. While those with PSP who do respond don’t need a long-acting form, avoiding having to take pills could be a major advantage for those with swallowing difficulty.
#8: DBS plus exercising may rewire Parkinson’s brain
The brain, even in older persons, can route its circuits around areas of damage, especially if those circuits are used often. That’s called “exercise” or “physical/occupational therapy” or just “practice,” (note my sarcasm) and it does work. This is true in PSP as well as in PD. But deep brain stimulation (DBS), at least the kind used in PD, does not work in PSP. That’s because the brain areas that are overactive in PD simmer down in response to DBS. But in PSP, those areas are part of the degenerative process itself, and are underactive. But to repeat: Informal physical activity and formal physical/occupational therapy do work in PSP. They should be an important part of the daily routine, assuming that one’s physician decides that any balance problems, osteoporosis, or cardio-pulmonary disorders are not contraindications.
#7: FDA approves bilateral Exablate Neuro treatment for Parkinson’s
Exablate is the brand name for a device that focuses destructive ultrasound beams on those over-active brain areas of PD. (It usually appears in the literature as “focused ultrasound” or FUS.) So, the idea is same as DBS, but FUS produces a permanent lesion that can’t be adjusted by the doctor based on the patient’s response. But its advantage of FUS over DBS is no hardware in the head, wires under the skin, pacemaker device in the chest, periodic battery changes, or explanations at metal detectors. Unfortunately, the FUS lesion locations used in PD would not work in PSP, but novel lesion locations could, in principle, be discovered for PSP for problems like balance, speech or cognition.
#6: Dosing starts in trial of anti-inflammatory therapy for Parkinson’s
PNT’s news item reporting the start of this single-center, Phase I trial appeared in March, as their blurb states, but I’ve got an update for you. The drug’s safety and tolerability were satisfactory but trial, with only 30 patient and one month’s observation, was not powered to demonstrate efficacy. The sponsor company, Ventus Therapeutics, has now started a Phase II trial for PD at 23 US sites. The drug, called VENT-02, targets NLRP3, a receptor protein activated by stress signals in the microglia, the brain’s equivalent of white blood cells. That could be relevant to PSP, where microglia help spread the abnormal tau protein through the brain and otherwise mediate the brain inflammation of PSP.
I’ll cover items #5 through #1 tomorrow or whenever my week’s chores permit.
Two terms used in the article that are probably not familiar to you in this context are “spasticity” and “apraxia.”
Spastic speech has a “rubber-band” quality, with abrupt variations in speed and loudness. It’s very common in PSP. It corresponds to spastic limb movements (rare in PSP), where joints can suddenly flex or extend in response to gentle movement or sensory input.
Apraxic speech features reduced ability to make certain sounds or words without a corresponding inability to form their components. It’s common in some of the APDs, but not in PSP. It corresponds to limb apraxia (common in PSP), where power and simple tasks are preserved, while more complex, learned tasks are impaired.
The paper includes videos of people with some of the APDs performing a standardized series of speech tasks, including describing the picture below:
The famous “cookie theft scene” test is designed to assess perception, judgement and language, but it serves as a test of voice and speech as well.
That brings us to the differences among “voice,” “speech” and “language.”
Voice difficulties include things like hoarseness and low/high volume.
Speech difficulties include things like slurring, slowing/speeding and disordered rhythm of sentences.
Language problems include things like wordfinding problems, word substitutions and reduced grammatical ability. It’s not discussed in the current paper but may be the topic of a subsequent review by the same group of neurologists.
A quick-and-easy way to organize this scheme is to consider voice problems as arising from the lungs and larynx; speech problems from the mouth, tongue and lips; and language problems from the brain.
The paper’s authors are all at institutions within CurePSP’s Centers of Care network. The leader of the project was Dr. Federico Rodriguez-Porcel of the Medical University of South Carolina. The other 25 authors, including yours truly, are listed in alphabetical order. So, as a clearly biased contributor, I recommend this paper on voice and speech in the APDs to those interested in understanding the range of such problems and their potential for rehabilitation.
Yesterday’s post was the first five of my top ten PSP news items of 2025. Here are the rest, again in approximate and subjective descending order of importance.
New ways of interpreting standard MRI images have gained ground as diagnostic markers for PSP. One is a test of iron content in brain cells called “quantitative susceptibility mapping” (QSM). Nine papers on that topic appeared in 2025, four in 2024 and none previously. It’s looking like combining QSM data from ordinary measurements of atrophy of PSP-related brain regions could be the ticket, as both measures come from the same test procedure, unpleasant though it may be, and they measure different things.
Positron emission tomography (PET) imaging of PSP’s type of tau (“4-repeat tau”) has made advances in 2025. This test requires intravenous injection of a “tracer” with a radioactive component that enters the brain tissue,sticks to the target molecule and is then imaged. It can distinguish PSP from non-PSP, distinguish among various PSP subtypes, and quantify the disease progression. The leading such tracer in terms of readiness for submission to the FDA is [18F]PI2620 and a distant second is [18F]APN-1607 ([18F]-PM-PBB3; Florzolotau). A tau PET tracer called Flortaucipir is on the market as a test for Alzheimer’s disease, but it performs poorly for PSP.
There’s brain inflammation in PSP, but it’s not clear whether it’s a cause or a result of the loss of brain cells, or both. Regardless, measuring the quantity and type of inflammation using blood or PET could shed light on the cause of the disease, identify new drug targets, and serve as a diagnostic marker. A good example of 2025 research on blood markers of inflammation in PSP is here and on PET imaging of inflammation is here .
We know of variants in 21 different genes, and counting, each of which subtly influences the risk of developing PSP or its age of onset. The area of the genome most important to PSP is the one that includes the gene encoding tau (called “MAPT”) on chromosome 17. The most important PSP genetic advance in 2025 was probably the discovery that some PSP risk is conferred by extra copies of a stretch of DNA, not the sequence itself. This news could inspire investigation of other places in the genome for other copy-number variants, which are much trickier to find than sequence variants. Here’s a great review of the latest in PSP genetics.
And lastly, a disappointment: a negative result of a double-blind trial of the combination of two drugs already approved for other conditions: sodium phenylbutyrate (“Buphenyl”) and taurursodeoxycholic acid (“TUDCA”). Blog post here.Sponsor’s press release here. Buphenyl protects the endoplasmic reticulum, which helps manufacture proteins, and TUDCA helps prevent brain cells from undergoing self-destruction (“apoptosis”) in response to various kinds of stressors. The pair were theorized to act synergistically. The trial’s upside is that its placebo group data can be used to provide better statistical support for future innovations in clinical trial design.
We saw some real progress against PSP in 2025. This post and the next list my top 10 developments from the past year in approximate and very subjective descending order of importance:
AZP-2006, a drug that enhances the breakdown of abnormal tau protein by the cells’ lysosomes, was found to slow the progression of PSP by 64%. The catch is that even that impressive-sounding result did not reach statistical significance because the trial was too small, having been designed mainly to assess safety. Furthermore, its design did not exclude the possibility of random bias in the selection of the participants. But the sponsor company plans to start a much larger and better trial in the second quarter of 2026 as part of the PSP Trial Platform. My blog post on that is here.
A new subtype of PSP has been identified and named PSP-PF. It was formed from chunks of two of the previously known ten subtypes, PSP-frontal and PSP-postural instability. If confirmed by other researchers, it will probably be the third-most common subtype, and the second-most rapidly progressive. The discovery could allow the expansion of drug treatment trials, which prefer to enroll rapidly-progressing participants, to include a more people with PSP than just the ~half with PSP-Richardson syndrome. My blog post is here.
CurePSP announced its new Biomarker Accelerator Program, offering grants of up to $500,000 for major projects to identify and characterize diagnostic tests for PSP. The program will consider applications involving not only markers to distinguish PSP from other disorders, but also those predicting an individual’s course and to assess change in the disease to use as outcome measures in treatment trials and other research.
Neurofilament light chain, a protein released into the spinal fluid and blood by many kinds of damage to brain cells, accumulated more evidence of its potential as a diagnostic marker of PSP. Although NfL is the most promising fluid marker for PSP, it’s not quite ready for routine use because of as yet insufficient sensitivity, specificity and consistency across different labs. Another major outstanding issue is to what extent blood can replace spinal fluid as a test medium. However, with the publication of each small advance, more research groups and funders become interested.
I’ve been thinking about PSP subtypes a lot lately, mostly because of last week’s report of an eleventh subtype, PSP-PF, comprising elements of the PSP-PI and PSP-F types. See my recent post for more explanation. I’ve read what I can about what causes the various subtypes to prefer slightly different parts of the brain. The general thought on that right now is “tau strains.”
Think of tau as a species, like the dog, and its strains as breeds. Let’s not get into the molecular nature of the inter-strain differences or what produces them. Instead, let’s recognize that those strains could theoretically underlie the differences among the 11 PSP subtypes by introducing differences in predilections for different groups of brain cells sharing a location or function. But this week, another possibility emerged as an explanation of the subtypes’ brain-area preferences: abnormal venous circulation.
The study in Parkinsonism and Related Disordersperformed brain MRIs and routine clinical office exams on 95 people with PSP. Of those, 64 had one of the three “cortical” subtypes (PSP-speech/language, PSP-corticobasal syndrome, and PSP-frontal). The other 31 had one of the “subcortical” subtypes (PSP-Parkinsonism, PSP-progressive gait freezing, PSP-postural instability). There were also 50 healthy participants as controls.
The three groups of participants were compared with regard to the size, number and location of any “white matter hyperintensities” (WMHs), examples of which appear in the MRIs below as the irregular white dots and splotches. In mild form, they’re common in healthy, older people and more so in those with high blood pressure, diabetes and other vascular risk factors. You can see how some of them sit smack up against the black slits in the middle of the brain, the spinal-fluid-filled lateral ventricles, and some are much closer to the outer, convoluted surfaces of the brain, the cerebral cortex. (Image from Inzitari D, Pracucci G, Poggesi, et al. BMJ. 2009 Jul 6;339:b2477. doi: 10.1136/bmj.b2477
The graph below shows the current paper’s main results: (from Fu M-H, Satoh R, Ali F, et al. Parkinsonism and Related Disorders. 2025 Dec 22:143:108170. doi: 10.1016/j.parkreldis.2025.108170).
This graph’s vertical axis is a measure of the WMHs’ total volume, expressed as a percentage of total intracranial volume. The horizontal axis is the WMHs’ location expressed as average distance from the lateral ventricle. The participants with the subcortical subtypes of PSP had the greatest volume of WMHs and their average distance from the lateral ventricles was greatest. The people with the cortical subtypes ranked lower in both measures, and the control participants ranked lowest. However, after correcting for various potential confounders, the differences remained statistically significant only for the areas between 12 and 30 mm from the lateral ventricles.
How to interpret this? Let’s start with some background:
The tiny veins draining blood from the brain are divided into deep and superficial systems. Each flows into its own set of larger veins en route to the heart.
WMHs are areas of scarring. They’re largely of unknown cause, but they correlate with risk factors for stroke, which is mostly related to narrowing of arteries, not veins.
Multiple sclerosis, which produces areas of white matter inflammation and scarring more severe than those of PSP, has been linked to insufficiency of venous drainage of the brain.
Normal-pressure hydrocephalus, which is similar in many ways to PSP and even can have PSP-like changes in the brain cells, has been shown (by the same research group as the present paper) to include insufficiency in one of the deep veins.
The areas of brain yielding the graph’s statistically significant results drain into the deep venous system. They’re unrelated to brain territories supplied by any specific arteries. The authors tentatively conclude that the WMHs may be caused by insufficiency in the brain’s deep venous system. They are appropriately cautious about assigning cause-and-effect, but the obvious question raised by their results is whether narrowing of the deep veins, and not any differences in tau or its post-translational modifications, could explain some, or maybe all, of the variety of PSP subtypes.
—
The authors of this paper overlap with those of last week’s about the new PSP-PF subtype summarized in my last post. All from the Mayo Clinic in Rochester or Jacksonville, they include first author Dr. Mu-Hui Fu working under senior author Dr. Jennifer Whitwell, a veteran leader in PSP-related imaging research.
As I occasionally do, today I’ll create a blog post by combining a reader’s comment on a previous post with my reply. This comment is from Jack Phillips, Chair of the Board of Directors of CurePSP:
Larry, with the more rapidly progressing PSP-PF and its large % of PSP patients, it seems to put even more urgency on our Biomarker Acceleration Program. Do you believe the biomarker program will be able to distinguish between the different subtypes of PSP? Jack
Hi, Jack,
Happy Holidays!
First, let’s assume that further research corroborates the existence, size, and statistical validity of the new subtype called PSP-PF, which isn’t a slam dunk.
You’re right that it would be great to have an accurate way to divide everyone with PSP into a) the two fastest-progressing types (PSP-RS and PSP-PF) and b) all the others. But first, let’s see if that can be done clinically (i.e., using good old history and neuro exam). Now that many of the people with the more aggressive variations of PSP-F and PSP-PI can be grouped as PSP-PF, the remaining, slower-progressing cases of PSP-F and PSP-PI would probably be easier to distinguish from PSP-RS than they were before, so maybe clinical would work well.
As for the ability of CurePSP’s pending biomarker program to do this job better than simple clinical evaluation: The first thing that comes to mind is to image the anatomical location(s) of the most intense tau deposition and/or inflammation in the brain. Second-generation tau PET using the tracers 18F-PI-2620 or 18F-APN-1607 can already do those things to an extent. That technique would now have to be refined and tested for its ability to identify PSP-PF.
So, yes, a PET marker to diagnose PSP-PF (or maybe a PSP-PF/PSP-RS group) is a realistic goal in the next couple of years. But the multi-million-dollar expense of all those experimental PET scans together with the administrative costs would be better handled by the companies making the PET ligands than by a relatively small nonprofit like CurePSP.
As a more affordable alternative to PET, markers of neurodegeneration intensity might be able to distinguish PSP-RS/PSP-PF from the more slowly-progressing PSP types. Measures of atrophy on ordinary MRI (conditioned on symptom duration at the time of the test) might be able to do this to an extent, as might serum levels of neurofilament light chain or inflammation-related compounds.
Perhaps an index combining those two with clinical history and exam could be the ticket. (Or perhaps all those tests would only succeed in identifying the same group of patients, in which case combining them would be pointless. But that would be good to know.) Now, that’s something that the CurePSP Biomarker Initiative could afford to fund.
You’ve all heard of the ten known PSP subtypes. They’re classified into three groups by the general areas of the brain involved (i.e., cortical vs. subcortical). Here’s the list:
Cortical and subcortical (~50% of all PSP)
PSP-Richardson syndrome (PSP-RS)
Cortical (~20%)
PSP-frontal (PSP-F)PSP-speech/language (PSP-SL)
PSP-corticobasal syndrome (PSP-CBS)
Subcortical (~30%)
PSP-Parkinsonism (PSP-P)
PSP-progressive gait freezing (PSP-PGF)
PSP-postural instability (PSP-PI)
PSP-ocular motor (PSP-OM)
PSP-cerebellar (PSP-C)
PSP-primary lateral sclerosis (PSP-PLS)
(As an aside: Neurodegenerative diseases are defined mostly by their pathological (i.e., microscopical) appearance, but each disease so defined may have several possible sets of outward signs and symptoms in the living person, depending on the general locations of the pathology within the brain. We deal with this by hyphenating the names of neurodegenerative diseases, with the pathology first and the clinical picture second.)
Now, to the news: Researchers at the Institute of Science in Tokyo, the Mayo Clinic, and UC San Diego have refined the above subtype classification. First author is Dr. Daisuke Ono, senior author Dr. Dennis Dickson and eight colleagues included 588 autopsy-proven cases of PSP from the Mayo brain bank without evidence of other neurodegenerative diseases. First, they used ChatGPT’s large-language software to extract clinical data from 53,527 pages of medical records, tabulating the order of appearance and progression rate of 12 pre-specified, PSP-related symptoms in each patient. Next, they performed a statistical technique standard for this sort of thing called “cluster analysis,” coupling it with a “decision tree model.” The first revealed groups of symptoms and progression rates that occurred together more often than would be expected by chance. The second worked out a practical, step-by-step way for neurologists to assign an individual patient to a subtype.
The most important result was a new subtype combining some patients with what has been defined as PSP-F with some from PSP-PI. The analysis still found statistical justification for continuing to recognize those two familiar categories as bona fide subtypes on their own. The new subtype, called PSP-PF (the continuous red curve), has the dubious distinction of having the most rapid progression and shortest total survival of all. In the graph below, you can trace a vertical line from where the “median” line crosses the curve for each subtype to find the median survival on the horizontal axis.
The median survival of PSP-PF was 6 years, with a 25-75 interquartile span (i.e., the middle two quarters of the group) of 5-7 years. This compares to PSP-RS, with a median of 7 and a 25-75 span of 6-8. For the subjects remaining in the PSP-PI and PSP-F groups, the median survival figures were 8 and 9 years, respectively.
This re-shuffling isn’t just a statistical detail. In the total group of 588 patients, 188 (32%) had PSP-PF, while only 68 (12%) had PSP-RS. Even considering the bias of any autopsy series toward over- representation of atypical cases, it’s still remarkable that PSP-PF is far more common than the other non-RS subtypes.
All that should be accompanied by the standard scientific conservatism about adopting new findings as textbook-worthy, especially without independent confirmation. Weaknesses in this study, all of which are recognized by the authors, include the following:
When a clinical feature wasn’t mentioned in the records, the analysis treats it as if it were known to be absent.
Quantitative data such as drug dosages, blood tests results, cognitive test results, and imaging details were not considered.
If a symptom onset date was not mentioned in the records, the date of the first relevant physician visit was used as the equivalent.
Having recognized all that, we can still say that an AI-based procedure may have found a pattern in ordinary medical record data that human neurologists and researchers missed.
My title for this post tentatively calls the discovery “unwelcome” only because no one would be glad to learn that their subtype of PSP is more rapidly progressive than they thought. (I’m referring to those people with PSP-PI and PSP-F who would fall within the definition of the new PSP-PF.) But one upside is that the news that a large group of people with PSP has a rate of progression similar to that of PSP-Richardson could allow neurologists to better counsel patients and their families. Another important upside is that perhaps clinical neuroprotection trials could now enroll participants with both PSP-PF and PSP-RS instead of confining themselves to the latter. This could greatly increase the pool of eligible trial participants and shorten the time required for the recruitment and double-blind periods.
The main potential obstacle to enrolling participants with PSP-PF into trials is that the primary outcome measure, the PSP Rating Scale, has not yet been validated for that subtype. But that should be possible to accomplish by identifying people with PSP-PF in existing, longitudinal or retrospective observational cohorts using the decision rubric of Ono et al. Then, one would simply assess the ability of their existing, longitudinally administered PSP Rating Scale scores to track their symptoms over time.
So the “unwelcome” part of this won’t actually change anyone’s PSP for the worse and the upside of speeding up clinical trials would be most welcome.
We still don’t have a great diagnostic test for PSP. The best we can do is about 80%-90% sensitivity, specificity and positive predictive value. In English:
Sensitivity is the fraction of people with PSP who give a positive result on the test.
Specificity is the fraction of people without PSP who give a negative result on the test.
Positive predictive value is the fraction of people with a positive test who actually have PSP.
A single number combining these into something useful in evaluating a single individual — rather than in comparing groups — is the “area under the receiver operating curve” (AUC; see this post for an explanation). The AUC ranges from 0.50, which is no better than a coin toss, to 1.00, which is perfect accuracy. An acceptable diagnostic test typically has an AUC of at least 0.85.
Most of the studies of PSP diagnostic markers have important weaknesses such as:
The studies frequently set up artificial situations such as distinguishing PSP only from PD or normal aging rather than from the long list of other possibilities that must be considered in the real world.
The patients’ “true diagnoses” are usually defined by history and examination alone rather than by autopsy.
The patients included in the study were already known to have PSP by history and exam (or sometimes by autopsy), while the purpose of the marker would be to identify PSP in its much earlier, equivocal stages or in borderline or atypical cases.
The patients with PSP in most such studies are only those with PSP-Richardson’s syndrome, who account for only about half of all PSP in the real world.
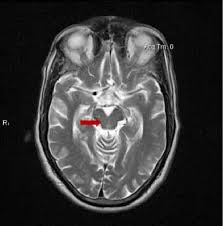
The best type of marker so far is ordinary MRI. Recently, a group of neurologists in Athens, Greece led by first author Dr. Vasilios C. Constantinides and senior author Dr. Leonidas Stefanis evaluated the specificity of various MRI-based measurements of brain atrophy. One strength of their study was that their 441 subjects included people not only with PSP and Parkinson’s disease, but also with a long list of other conditions with which PSP is sometimes confused as well as a group of healthy age-matched controls.
The single best MRI marker per this study was the area of the midbrain, the fat, V-shaped structure indicated below:
They found that MRI markers provided:
High diagnostic value (AUC >0.950 and/or sensitivity and specificity ∼90 %) to distinguish PSP from multiple system atrophy, Parkinson’s disease, and control groups.
Intermediate diagnostic value (AUC 0.900 to 0.950 and/or sensitivity and specificity 80 % to 90 %) to distinguish PSP from Alzheimer’s disease, frontotemporal dementia, dementia with Lewy bodies, and mild cognitive impairment (an early stage usually of AD).
Insufficient diagnostic value (AUC < 0.900 or sensitivity/specificity ∼80 %) to distinguish PSP from corticobasal degeneration, normal-pressure hydrocephalus, and primary progressive aphasia (a language abnormality that can be caused by multiple specific diseases).
Insufficient value to distinguish the non-Richardson PSP subtypes from corticobasal degeneration and primary progressive aphasia, but good performance in the other comparators.
The researchers also concluded that:
One MRI measurement isn’t best for all the possible PSP comparators.
Sometimes a combination of two or three measurements performed better than any single measurement.
One weakness of their method was the use of subjects diagnosed by standard history/exam (i.e., “clinical”) criteria, rather than by autopsy. Another is that their patients with PSP had had symptoms for an average of three years, so these were not subtle or early-stage cases. A letter to the journal’s editor from Dr. Bing Chen of Qingdao City, China further pointed out that the study of Constantinides and colleagues failed to account for the subtle effects of neurological medications on brain atrophy. As PSP and the comparator disorders may be treated with different sets of drugs, taking this factor into account might enhance or reduce the apparent diagnostic value of MRI atrophy measurements.
So, bottom line? Drs. Constantinides and colleagues have given us the first study of MRI markers in PSP to include meaningful numbers of subjects with non-Richardson subtypes. It’s also one of the few studies of any kind of PSP marker to include comparison of PSP a wide range of diagnostic “competitors” beyond just Parkinson’s and healthy aged persons. Another plus is that the test, routine MRI, is nearly universally available, relatively inexpensive, and non-invasive.
The hope is that Pharma companies or others with candidate drugs will now have fewer or lower hurdles in the way of initiating clinical trials.