Another step forward: The FDA, for the first time, granted permission for a tau PET ligand to proceed to a Phase 3 clinical trial. What does that mean?
A compound called “fluorine-18 APN-1607” (18F-APN-1607) is being developed as a PET ligand for PSP because of its affinity for the tau protein. It specifically targets 4-repeat tau, the type found in the neurofibrillary tangles of PSP and corticobasal degeneration. The ligand, which we’ll call simply “1607,” has passed safety testing and has been found able to distinguish PSP from its most likely diagnostic alternatives: Parkinson’s disease and the Parkinsonian variant of multiple system atrophy. Of course, both of those are alpha-synucleinopathies, not tauopathies. But 1607 can also distinguish PSP-RS from the two next-most-common kinds of PSP, called PSP-Parkinsonism and PSP-progressive gait freezing, and from corticobasal degeneration.
Now the challenge is to see if 1607 can do those things better than a good neurologist, which means also a useful degree of accuracy in the early, diagnostically equivocal stages, not just after the disease is well-established, as in the trials to date . The FDA-approved plan for the new trial will administer the scan to people with “possible” PSP, wait up to 18 months, and then compare those with positive scans to those with negative scans with regard to whether they’ve progressed to “probable” PSP. The formal PSP criteria published in 2017 provide formal definitions of “possible” and “probable” PSP. “Definite” PSP requires autopsy, and as part of its statistical analysis, the trial will analyze any patients, hopefully a minority, who come to autopsy during the 18-month period.
How long will this all require? It will probably take a year to recruit all the patients, and the last patient will finish 18 months later. Add a few months at the start for administrative issues and a few more at the end for data analysis, and we’re talking about 3 years. But it’s progress, and the FDA has indicated that they would consider approving the product on the basis of this single Phase 3 trial, assuming the results are convincing. For most drugs, the FDA requires more than one Phase 3 trial.
Disclosure: The drug company behind 1607 is called Aprinoia Therapeutics, based in Cambridge, Massachusetts. I serve as a paid consultant for them, advising in design of their trial. I have no stock in Aprinoia or other financial interest in the success or failure of 1607. In that spirit, I’ll also mention that another tau PET ligand, 18PI-2620, sponsored by a German company called Life Molecular Imaging, is close behind 1607 in the development pipeline and is showing similar utility.
In case you missed the front-page story three days ago (3/8/24) in The New York Times, the drug Relyvrio has failed to show benefit in a large (664 participants), Phase 3 trial in amyotrophic lateral sclerosis. The drug did appear to show benefit in a much smaller (137 participants) Phase 2 ALS trial in 2020 and was provisionally approved for sale by the FDA on the strength of that result. Now, the drug company, Amylyx, may have to discontinue marketing the drug for ALS.
Why is this relevant to PSP? Because four months ago Amylyx initiated a 600-participant Phase 3 trial of Relyvrio for PSP. It’s called “ORION.” So far, recruitment has begun at only a handful of sites, all in the US, with plans to expand into Europe and Japan over the coming months. The FDA’s permission to start ORION without a Phase 1 or 2 in PSP was based in part on the success of the drug in the Phase 2 ALS trial. See my post of February 29 for details.
Amylyx is also testing Relyvrio in people with Alzheimer’s disease, where a Phase 2 trial has demonstrated adequate safety and tolerability. I have no information on a Phase 3 in AD.
The question now is whether the ORION trial in PSP will continue. So far, there’s been only one business day since the ALS news, and I’m not sure if the top brass at Amylyx — or the company’s sources of financing — have yet decided. But the Times article reported that the FDA approved Relyvrio for ALS only after Amylyx agreed to withdraw the drug if the Phase 3 trial showed no benefit. Furthermore, right after the ALS trial news hit, the stock price of Amylyx dropped from $19 to $3 and stayed about there. Stock markets usually know how this sort of news is likely to play out. Relyvrio is Amylyx’s only marketed product but they do have other drugs in the development pipeline.
The mechanism of action of Relyvrio addresses issues important to both disorders, which suggests that if it failed in one, it could well fail in the other. But we don’t really understand the pathogenesis of either disease well enough to know if PSP might respond when ALS did not.
Meanwhile, if you were planning to try to enroll in the ORION trial, I’d advise you not to change your plans. The ALS trial showed no important toxicity, at least in people with ALS, and you wouldn’t want to lose your potential spot at the study site because you delayed enrolling until definite information on the future of the ORION trial became available. When other trials in PSP start enrolling, that advice could change, of course.
(I’ll repeat the disclosure I made in my 3/8/24 post: I’m a paid consultant for Amylyx, advising them on design of the ORION trial and training the participating neurologists on proper administration of the PSP Rating Scale. But I have no stock in the company nor other financial interest in the drug’s success.)
I have good news and bad news, and they’re the same piece of news.
You’ve heard of Huntington’s disease. That’s the strongly hereditary condition that causes dementia and involuntary movements, starting at an average age of about 40 and leading to severe disability and dependency within 15 years or so. Its most famous victim is Woody Guthrie, the folksinger/songwriter who died in 1967. HD caused by a pathogenic variant (the word “mutation” is no longer considered appropriate) in the gene encoding the protein “huntingtin,” rendering it toxic to brain cells.
The variant in HD is a “trinucleotide repeat expansion.” The gene has a region where the three nucleotides cytosine, adenine and guanine (CAG) are repeated multiple times. The normal number of such repeats is less than 27. Everyone with 40 or more repeats will develop HD and each of their children has a 50% chance of inheriting the abnormal version of the gene. For 28 to 39 repeats, the person usually remains healthy, but the number of expansions can increase during the process of producing sperm cells or ova, putting subsequent generations at risk. A number of other neurological diseases are caused by CAG expansions in different genes.
Huntingtin protein has several known functions in the brain, but the damage to brain cells probably is not the result of loss of any of those. Rather, the excess number of CAG repeats encodes a an excessively long string of the amino acid glutamines. That gives the protein its toxic property, but we’re still not sure how it works or what to do about it. We do know, though, that in HD, brain cells have clumps of strands of glutamine, just as in PSP there are clumps of tau protein.
In PSP, there is no single culprit gene. Unlike HD, PSP only very rarely runs in families and each of the 14 genetic alterations currently known to be associated with PSP confers only a slight degree of risk. I know this looks cryptic, but here’s the current list of PSP-related genes in approximate order of discovery date: MAPT, STX6, MOBP, EIF2AK3, SLCO1A2 (two loci), DUSP10, RUNX2, LRRK2, APOE2/2 genotype, CXCR4, EGFR, GLDC, and C4A.
To this list we can now add “HTT,” the Huntington’s gene. That’s because last month, researchers announced that they had sequenced HTT from 588 people with autopsy-proven tauopathies, including 98 with PSP, along with controls without neurodegenerative disease. They found that while only 0.2% of the controls had at least 27 CAG repeats, for the people with PSP, the figure was 3.2% and for those with corticobasal degeneration, 2.7%. Keep in mind that in only a small fraction of those 3.2% will the number of CAG repeats expand far enough into the toxic range to pose a risk to the children or grandchildren.
The paper’s first author is Dr. Sergio Pérez-Oliveira and the senior author is Dr. Victoria Álvarez, both geneticists at Hospital Universitario Central de Asturias, in Oviedo, Spain, along with 21 other collaborators. The paper appeared in Brain Pathology, a well-respected journal.
So, the bad news I mentioned is a very slight increased risk of HD occurring in future generations of families with a member with PSP. What’s the good news? It’s that we now know of another genetic risk factor for PSP, and it’s one that we already knew a lot about, thanks to decades of research on HD. We can compare the long list of known actions of huntingtin in the brain to the long list of actions of the 14 other PSP-related genes. More important, we can compare the known toxic action of the excessively long strings of glutamine with the list of ways in which brain cells are damaged in PSP and look there for synergistic interactions and for drug targets to disrupt such processes.
Overall, I’d say that the good news definitely outweighs the bad news, first because statistically, the bad news is only a very small risk, and the new publication doesn’t change it – it only reveals it. But the revelation of the good news gives PSP researchers something new to sink their teeth into in their search for a prevention and cure.
Yesterday a reader left a comment regarding my 2/29/24 post on the ORION trial (of the drug AMX-0035) and I responded on the comments page. But I thought the comment was so well expressed and possibly so widely shared by my readers that it deserved more of a platform. So I turned the question and my response into this post.
Hello Dr. Golbe,
Thank you for the information and data on the AMX-0035 trial. I cannot help but share my thoughts.
The endpoint of this trial, if I understand correctly, is to slow the progression of the disease’s natural course. How is this to be assessed via the PSPRS? An absence of a rise in the score over time or an improvement in the score? If the goal is to prevent a rise in the score over time, is there a known rate at which the PSP-RS usually rises in the absence of any intervention in order to compare this to?
I hesitate to bring this next concern up, however I feel that I need to. In the absence of any known treatment for PSP, if this trial is successful then this is quite good news – anything that helps in any way is good news. However, is it really? The quality of life for a patient with PSP is terrible as you (or anyone who has ever cared for or evaluated a patient or loved one with PSP) know. Therefore is extending this poor quality of life by a year truly a success? The reason I bring this up is because I would like to know if perhaps by targeting these molecular and cellular processes within the mitochondria and endoplasmic reticulum, and thus reducing the stress and burden which is on these patients at the cellular level, is there any hope that perhaps there will also be some symptom improvement as a result of a lessening of burden/overload of the system and the brain’s own immune system and other processes being able to more efficiently function or discard of more abnormal proteins? And therefore some (even small) improvement in quality of life. This very well may be completely unknown. I may have asked you this in a prior post. I ask this not to be dismal or morbid but to see if there is even more hope. As you always say, hope is important.
AF
Dear Ms. F,
I think both of your questions are shared by many others.
The answer to you first question, regarding what the patients on the study drug are compared to, is the placebo group. At the time of enrollment, each patient is randomly assigned to receive either the real study drug or an identical-appearing placebo. Of course, this plan is made quite clear in the informed consent process but only the drug company knows the assignments. For the ORION study, 60% will get real drug and 40% placebo. At the end of the study, the rates of progression of the PSPRS score for the two groups are compared. In this way, we don’t need to know in advance how rapidly PSP progresses. On completing their 12 months of placebo or real drug, each patient will be offered the chance to take the real drug (called the “open-label phase”).
The second question asks about the likelihood of symptomatic benefit, rather than just slowing the rate of worsening. The chance of that is low, but not zero. If the drugs do improve the function of cells affected by the PSP process, some of them may be able to recover but others (probably most) will be beyond saving. Either way, the treatment may allow cells that are still healthy to avoid becoming involved in the disease process at all, or with a major delay. What we don’t yet have is a way to restore the function of the cells already lost, though researchers are busily working on that.
Your second question implies that it may not be worthwhile merely to slow the progression of a disabling condition without curing it or improving its symptoms relative to the study baseline. That’s a legitimate philosophical and ethical question. My own interactions with patients in my decades of practice and my informal poll of this blog’s readers show that even a 25% slowing of progression (the benefit of AMX-0035 in ALS), which would provide one more year of life for people with PSP, would be worth the hassle and risk of side effects of a new drug. Keep in mind that in prolonging survival from three years to four, the drug would not merely prolong the most advanced, disabled stage for another year. Rather, it would prolong the condition in each of the three years by 33%.
I hope you will forgive my 24-day posting hiatus. To make it up to you, I bring good news: The trial of AMX-0035 in PSP is planning to expand its enrollment activities in the next few weeks, and this drug’s track record is unusually encouraging.
The trial, dubbed “ORION” for some reason, initiated enrollment over the past two months at eight sites in California, Florida, Massachusetts, Michigan, Tennessee and Texas. 32 other sites in the US and dozens in Europe and Japan will open in coming months, with a total enrollment target of 600 patients. Those interested can email clinicaltrials@amylyx.com, check clinicaltrials.gov or the company’s own site. The trial will include a 12-month double-blind period with a 40% chance of assignment to the placebo group, followed by a 12-month open-label period. Trials like this usually take about a year or two to fully enroll, another year for the last enrolled participant to complete the double-blind and another few months to analyze the data.
The drug company is Amylyx Pharmaceuticals, based in Cambridge, Massachusetts. They held a meeting a few days ago for their US sites’ neurologists and coordinators, where I gave a detailed lesson on proper administration of the PSP Rating Scale, which will be the study’s main outcome measure. (Disclosure: Amylyx paid me for that presentation and for general advice on the trial’s design but I have no financial interest in the success of the company or the drug.)
The treatment in question is actually two drugs, taurursodiol and sodium phenylbutyrate, both administered orally as a powder stirred into water. The first addresses the dysfunction of the mitochondria in PSP. The second reduces stress in the endoplasmic reticulum and enhances the unfolded protein response, both of which are also dysfunctional in PSP. All of these cellular functions are related and lab experiments show that the two drugs combined work better than the sum of their individual effects.
Unlike any of the other new drugs currently or or soon to be tested for PSP, AMX-0035 has been found to help a related disease, amyotrophic lateral sclerosis (ALS or Lou Gehrig disease), where it appears to slow the progression by about 25% and prolongs survival accordingly. The drug, branded “Relyvrio,” won approval from the FDA for ALS last year and is gaining widespread acceptance among neurologists in treating that condition.
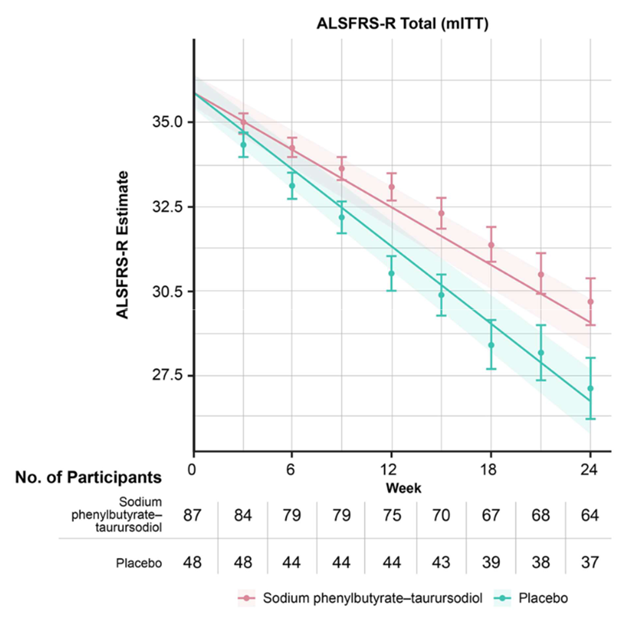
The graph below (from Paganoni et al, New England Journal of Medicine, 2020) shows the worsening of the main ALS disability measure (vertical axis; note that the bottom is not zero) over the 24 weeks of the trial (horizontal axis). The orange line/shaded areas and the means/standard error bars represent the patients on AMX-0035 using two different statistical techniques. The patients on placebo are shown in green.
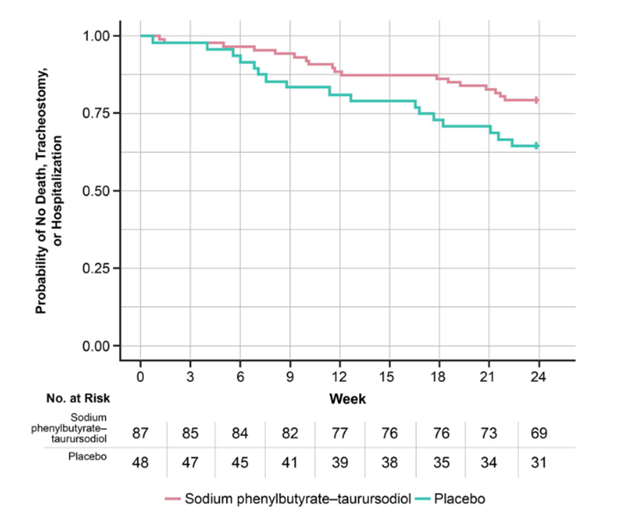
The graph below (also from Paganoni et al), called a Kaplan-Meier survival plot, shows the fraction of patients in the ALS trial remaining alive without tracheostomy or hospitalization (vertical axis) along the 24 weeks of the trial (horizontal axis).
This is great for people with ALS, but that’s not a tau-based disorder like PSP. However, in a Phase 2 trial in Alzheimer’s disease, which is partly a tau disorder, AMX-0035 did reduce spinal fluid levels of both total tau and of a toxic form called p-tau 181. That trial was too small and brief to reveal any efficacy of AMX-0035 to slow or halt AD progression but I assume a proper Phase 3 trial will follow.
Side effects of AMX-0035 in the AD trial have not been published, but in the ALS trial, nausea, diarrhea, excess salivation, fatigue and dizziness occurred in 10% to 21% of patients on the drug and in slightly lesser percentages of those on placebo.
If AMX-0035 shows the same result in PSP as it did in ALS, that means about one additional year of survival for the average patient, and even more if the disease can be diagnosed earlier. Potential game-changer. I’ll keep you updated.
“Pheno-“ is the Greek root for “outward appearance” and so far, PSP has ten of them. The differences arise from varying emphasis of the degenerative process among different parts of brain. While standard laboratory methods show that at the cellular level the pathology among the ten phenotypes is identical, a few details are starting to emerge using more recent and sophisticated techniques at the molecular level.
The most common PSP phenotype, called PSP-Richardson’s syndrome (PSP-RS), is the one Steele, Richardson and Olszewski originally described in 1964. The others were published piecemeal starting in the early 2000s. J.C. Richardson was the leader of the trio at the University of Toronto, a senior clinical neurologist who noticed an unusual form of Parkinsonism among his patients in the 1950s and 60s. John C. Steele was his trainee and Jerzy Olszewski was the neuropathologist who described the corresponding microscopical abnormalities. So, it’s altogether fitting and proper that Dr. Richardson should be honored in this way.
Prevalence of the phenotypes. The percentage of the PSP population with each phenotype has not been studied in a true community-based population. The published percentages vary widely across centers and are all from referral-based populations at research institutions, where unusual forms of diseases are over-represented to varying degrees. Even without that issue, several things make it hard to be sure of the prevalence of the various phenotypes:
It’s difficult to estimate the population prevalence of atypical cases of PSP from autopsy series because atypical cases are more likely to come to autopsy, and without autopsy, it’s hard to know that someone with atypical PSP really had PSP.
In their later years, all of the phenotypes tend to merge into a PSP-RS appearance, so the relative frequencies of the phenotypes may depend on the patients’ disease stage when the researchers evaluated them.
Clear diagnostic criteria do not yet exist for many of the phenotypes and many patients satisfy criteria for more than one, even in early stages. A method has been published for how to deal with this, but most of the publications antedate or ignore it.
Although the original differentiation of PSP-RS from PSP-P in 2005 did use a rigorous statistical technique called “factor analysis” to confirm that the two are distinct, this is not usually the case for the other phenotypes relative to PSP-RS or to one another.
Nevertheless, here are my very rough estimates of their contributions to PSP in general, based on a Gestalt impression of the literature:
Richardson’s syndrome
45%
PSP-RS
Parkinsonism
25%
PSP-P
Frontal
10%
PSP-F
Progressive gait freezing
5%
PSP-PGF
Speech/language
5%
PSP-SL
Corticobasal syndrome
3%
PSP-CBS
Postural instability
3%
PSP-PI
Ocular motor
3%
PSP-OM
Cerebellar
<1%
PSP-C
Primary lateral sclerosis
<1%
PSP-PLS
Many recent research articles group these into three categories based on their anatomical predilections in the brain: cortical vs subcortical. PSP-RS falls into neither of these because it has approximately equal degrees of both cortical and subcortical features. One important, practical reason for the grouping of phenotypes is to have groups large enough for meaningful statistical analysis.
PSP-Parkinsonism. The most common “atypical” (i.e., non-PSP-RS) phenotype of PSP is PSP-Parkinsonism (PSP-P). Relative to PSP-RS, it features more asymmetry, generalized bradykinesia, tremor, and levodopa responsiveness, and only later displays falls and cognitive loss. It is usually initially misdiagnosed as Parkinson’s disease. It has perhaps the slowest course among the PSP phenotypes, averaging about 9 years’ survival from symptom onset. This compares with about 6 years for PSP-RS and intermediate figures for the other phenotypes. In fact, the PSP-subcortical group as a whole has a similarly longer average survival duration than the PSP-cortical group as a whole or the PSP-RS + PSP-cortical groups.
PSP-progressive gait freezing. After PSP-P, the most common atypical phenotype is PSP-progressive gait freezing (PSP-PGF). In fact, most patients exhibiting only progressive gait freezing will eventually develop diagnostic features of PSP. The central feature of PSP-PGF is loss of ability to continue ongoing gait, especially after a pause, during a turn, or at a doorway threshold. In advanced cases, the patient cannot initiate gait at all. The picture also includes rapid, small handwriting and rapid, soft speech as frequent or severe features. The anatomic location of the pathology in such cases differs from that of PSP-RS in showing less involvement of the base of the pons (part of the brainstem) and of the dentate nuclei (part of the cerebellum).
PSP-speech/language. This is a composite category. In PSP-nonfluent/agrammatic variant of primary progressive aphasia (PSP-nfaPPA) speech is halting, with poor grammar, syntax, and pronunciation, but with normal comprehension and naming. A mirror-image variant called semantic-variant primary progressive aphasia (svPPA) features difficulty in naming with reduced vocabulary but with normal grammar and syntax. Together, PSP-svPPA and PSP-nfaPPA are referred to as PSP-speech/language disorder (PSP-SL).
PSP-corticobasal syndrome. CBS as a clinical syndrome (meaning a group of signs and symptoms that occur together, although the underlying disease may differ across patients) comprises highly asymmetric rigidity, slowed movement, and apraxia (loss of skilled movement), often with equally asymmetric dystonia (fixed postures), pyramidal findings (weakness and abnormal reflexes), myoclonus (small, rapid, irregular movements), and cortical sensory signs such as astereognosis (inability to identify objects by feeling them) and agraphesthesia (inability to identify figures traced on the skin). Aphasia (difficulty processing language) and other abnormalities localized to specific brain areas may also occur. Dysarthria (difficulty with pronunciation) can be prominent but gaze palsy, postural instability, and cognitive loss tend to be later and milder than in PSP-RS.
PSP-frontal. More formally called PSP-behavioral variant frontotemporal dementia (PSP-bvFTD or simply PSP-frontal), this phenotype features disinhibition, irritability, apathy, and loss of empathy for others, along with impairment in frontal “executive” functions such as ability to maintain attention, to follow instructions, to shift tasks on command, and to inhibit an ongoing action when appropriate. This is the core of the cognitive and behavioral deficits in PSP-RS, but when it appears first and remains worst, the term PSP-F is appropriate.
PSP-ocular motor and PSP-postural instability. Perhaps unsurprisingly given the cardinal features of PSP-RS, PSP can also take the form of a relatively pure ocular motor picture or a relatively pure picture of severe postural instability with falls and little else to suggest PSP. However, reported cases are very sparse to date. These have been designated PSP-ocular motor (PSP-OM) and PSP-postural instability (PSP-PI).
PSP-primary lateral sclerosis. The pathology of PSP can also produce the clinical picture of primary lateral sclerosis. PLS is one of the phenotypes of amyotrophic lateral sclerosis (ALS; Lou Gehrig disease) and can be difficult to distinguish from it, especially as ALS can produce frontal cognitive difficulties in many cases. The clinical picture of PSP-PLS is highly asymmetric and resembles that of CBS but with little or no cortical sensory loss (spatial sensation ability), dystonia (fixed postures), or myoclonus (very quick, small, irregular involuntary movements).
PSP-cerebellar. The classic lurching gait of PSP-RS has a cerebellar appearance, the speech of PSP has an ataxic (or drunken-sounding) component in many cases, and the ocular square-wave jerks of PSP occur commonly in cerebellar disease. For decades, these were considered minor and inconsistent features, but in 2009, neurologists in Japan described a PSP phenotype of PSP with involvement of the cerebellum at autopsy and early, prominent ataxia of the trunk and limbs. Although that original report found PSP-C in 14% of all people PSP at a Japanese center, the figure is much lower in Western populations for unclear reasons.
A word about drug trial eligibility. Until the cause of PSP in general is better understood, neuroprotection trials — those aimed at the fundamental brain cell loss rather than merely at ameliorating the symptoms — will continue to recruit only patients with PSP-RS. Why?
We don’t yet know if the non-PSP-RS phenotypes share – either with PSP-PS or with one another — the same molecular abnormality being targeted by the drug.
While the classic PSP pathology underlies close to 100% of PSP-RS, that figure is much lower for some of the other nine phenotypes. That means that non-PSP-RS may well be a non-PSP pathology, and that admitting participants with non-PSP-RS to a drug study runs a risk that some may not have PSP at all. This could obscure any benefit the drug may have unless the trial is prohibitively large.
The main outcome measure for nearly all PSP trials is the PSP Rating Scale, which was designed for, has been validated for, PSP-Richardson’s syndrome alone.
Trials to slow the progression of PSP use the rate of progression as the “outcome variable” of the trial. As noted above, the phenotypes do vary in their expected survival durations, and by inference, their progression rates. Therefore, including phenotypes with different inherent rates of progression would require a larger, longer and more expensive trial. One solution would be to make sure the active drug group and the placebo group receive similar proportions of the various phenotypes (a time-proven technique in trial design called “pseudo-randomization”). But this doesn’t solve the four preceding problems.
As PSP-RS progresses faster than the other nine phenotypes, a trial enrolling only that phenotype can reach a result in a shorter time or with fewer participants. This isn’t only financially advantageous for the trial’s sponsor. If the drug is ineffective or harmful, fewer patients will have been exposed to it, and if it’s effective and safe, it will reach market approval more quickly.
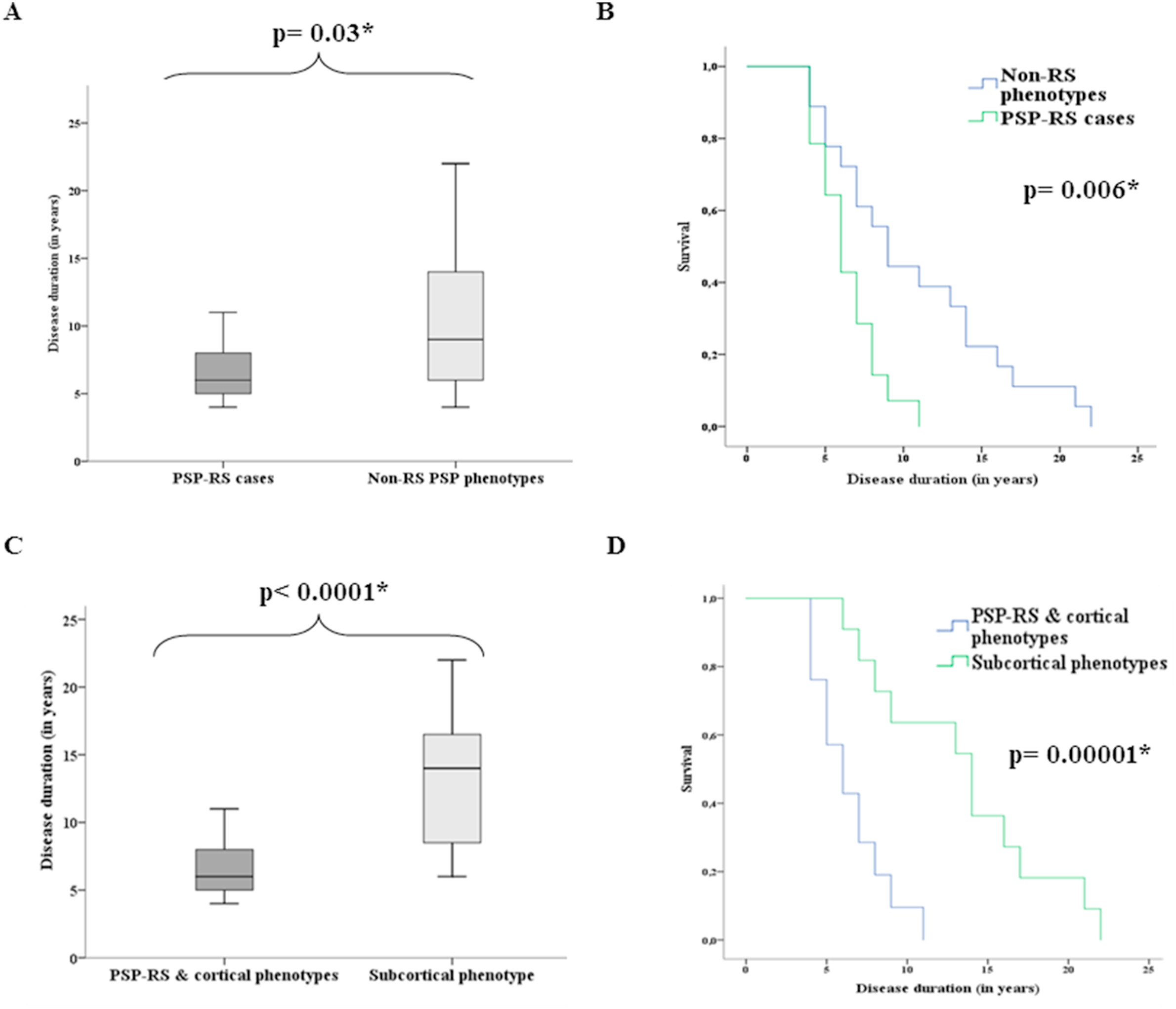
These graphs are from a review of records of 32 people with autopsy-proven PSP in Spain and Germany. The lead author was Dr. Mar Guasp of Hospital Clínic de Barcelona and the senior author was Dr. Yaroslau Compta of the same department. They show the difference in survival from initial symptom to death between the PSP-RS and non-PSP-RS (top) and between [PSP-RS + PSP] and PSP-cortical (bottom). Graphs A and C: For each box, the horizontal line is the median, the upper and lower borders are the 25th and 75th percentiles and the ends of the “whiskers” are the highest and lowest values. Graphs B and D show the same thing in the form of “survival curves” or “Kaplan-Meier curves.” The vertical axis shows the fraction of the original patients still living at the time (post-onset) shown on the horizontal axis. The p values are the likelihood that the difference could have happened by random chance. The asterisk indicates that this likelihood is low enough for the difference between the groups to be considered “statistically significant.” (Statistical veterans: Sorry to belabor this for the benefit of the statistical novices.)
To-do list:
Let’s figure out why the disease spreads through the brain in PSP-RS and the other PSP-cortical phenotypes more quickly than in PSP-subcortical. Efforts to do that have in fact begun and could provide the key to the whole puzzle of PSP.
Let’s agree on a way to enroll people with non-PSP-RS phenotypes into clinical trials. Current efforts to diagnose PSP using tau-based positron emission tomography (PET) and measures of tau in skin, blood or spinal fluid could potentially identify people with PSP other than PSP-RS who could potentially join a trial.
Let’s educate neurologists to identify, or at least to suspect, the non-PSP-RS phenotypes. This would allow them to avoid or delay fruitless diagnostic testing and to provide their patients with useful prognostic information.
The first of a raft of experimental PSP drugs in the pipeline has begun enrollment in a large treatment trial.
The ORION trial (don’t ask me what the acronym stands for) is sponsored by Amylyx Pharmaceuticals. Their press release is here. A bit more information is at the company’s website. Their email address is clinicaltrials@amylyx.com.
Clinicaltrials.gov lists more info, including a phone number (in the UK): +44 (808) 1642604. Only a few of the planned 33 sites in the US are open so far and no sites outside of the US has yet opened, to my knowledge. Those will be in Europe, Canada and Japan.
The product is actually two drugs called taurursodiol and sodium phenylbutyrate. They have a variety of beneficial actions in brain cells affected by PSP, but it’s not known which would be most important. Both drugs are already on the market, the first over-the-counter and the second with prescription. Neither alone is intended for brain diseases and each alone has only a minimal effect on the brain. But the two together have a synergistic effect on the brain and spinal cord that has proven modestly beneficial in ALS and has already received FDA approval for that disorder under the brand name Relyvrio.
There’s a lot more you’d want to know about the study’s design and the participants’ obligations, so please read the material at the links I’ve provided above and contact the company directly.
Amylyx has hired a contract research organization called Cronos to actually run the trial. They’re a subsidiary of a company called IQVIA. So if you encounter those names, no worries.
One more little thing: full disclosure. I’m a paid consultant for Amylyx, but that’s only for advice in the design of the trial and instructing the examining doctors how to use the PSP Rating Scale. They don’t pay me to recruit patients and my fee does not depend on the trial’s enrollment. Nor do I own stock in the company or have any other financial incentive to see the drug succeed. (Of course, I do have an emotional incentive for that — no secret there.)
This week, our knowledge of the genetics of PSP has more than doubled. First, as usual, some background:
Like many other complex conditions like atherosclerosis, schizophrenia and most cancers, PSP does run in families a bit more often than expected by chance. But as in those diseases, the familial tendency is too weak to produce the classic dominant or recessive pattern associated with a single, strongly-acting gene variant as in Huntington’s, Tay-Sachs or sickle cell anemia. Besides, adding up the risks from the known PSP-related genes wouldn’t explain the incidence of the disease in the population, rare though it is. That has prompted the theory that some unidentified external exposure or experience also has to play a role.
Over the past 25 years or so, a number of gene variants have been found to confer slight risks for developing PSP. The first-discovered and still the most important, called the “H1 haplotype,” is a complex set of variants in region of chromosome 17 that includes MAPT, the gene encoding the tau protein. Another four variants on other chromosomes were published in 2011 by CurePSP’s PSP Genetics Consortium.
In the years since, nine other variants were added piecemeal by other researchers. Those first 14 were all discovered using a technique called “marker association,” which only identifies a region of about 100 genes where the culprit gene would be located. The gene from those 100 that’s reported as a “hit” is generally the one with the best statistical association with the marker along with a scientifically rational reason to be associated with the disease under study. A more finely-grained search would actually work out the sequence of the genetic code, comparing people with PSP to those without PSP. That wasn’t practical back in 2011, but now it is. It’s called “whole-genome sequencing” or WGS.
The new list of gene variants has been found by an international WGS collaboration that grew out of the original CurePSP-supported team. They used DNA samples from 1,718 people with PSP, of whom 1,441 were autopsy-confirmed, and 2,944 samples from people without PSP as controls. The leaders are at the University of Pennsylvania and UCLA, but 26 other research institutions in nine countries contributed.
They confirmed five of the six previously-identified variants (the sixth came very close) and added seven new ones. They also elucidated new details of the cluster of variants in the H1 region. Most remarkably, they confirmed a previous, smaller study showing that PSP reverses the relationship of Alzheimer’s disease with the ApoE gene on chromosome 19. In AD, the epsilon 4 variant of ApoE is over-represented relative to controls and the epsilon-2 variant is under-represented, while in PSP, it turns out that those proportions are reversed despite the fact that both AD and PSP are tauopathies.
So far, the research article is only posted on medRxiv (“med archive”), a website for manuscripts not yet through the peer review process at a journal. (But my brain’s blogging center couldn’t restrain itself.) The next steps for the authorship team are to gather online comments on the manuscript from other scientists and to submit the resulting revision to a regular journal. There, the peer review may dictate other changes. The next scientific step will be to figure out what the mutations are doing wrong, determine to what extent the variants increase or decrease the amount of the protein they encode (called “expression studies”), and look for proteins encoded by those genes (or for proteins they interact with) that might be modulated by drugs.
As far as I can tell, even the newly expanded list of risk variants doesn’t explain enough of the overall cause of PSP to be used as a diagnostic panel. But it’s a start in that direction.
My canned lecture on PSP includes a slide on the two dozen or so most important scientific milestones in PSP research since the disease was first described in 1963. This paper is going there. As I learn more about the publication progress and clinical implications of this work, I’ll keep you all apprised.
Decades ago, the discovery that specific proteins aggregated in the brain cells of specific neurodegenerative diseases was a major advance. But like so many other scientific breakthroughs, it created another question: Why are there so many different clinical pictures among different people with the same neurodegenerative disease (like PSP) despite the fact that they all host the same aggregating protein (in this case, tau)? The ability of abnormal tau to “seed” the disease process into previously healthy brain areas is at the root of the disease process, but we’ve had scant clue as to how that works, exactly.
For PSP, the most important clinical variable is the eight subtypes (PSP-Richardson’s syndrome vs PSP-Parkinsonism vs PSP-progressive gait freezing, etc), and slightly less variable features are the onset age and rate of progression. In the past year or two, it’s become clear that the different subtypes tend to emphasize different areas of the brain, but that doesn’t explain why two people with the same subtype can have different onset ages and rates of progression.
This mystery became even more mysterious recently when a new electron microscopy technique called “cryo-EM” proved able to visualize individual protein molecules. It showed that for everyone with a given disease, the protein for that disease had the same misfolded shape. In other words, the tau molecule assumes the same rigid squiggle in everyone with PSP, a different rigid squiggle in everyone with Alzheimer’s, yet another in everyone with corticobasal degeneration, and so on. But that raised the question as to the reason for the variability among patients of the PSP onset age and rate of progression.
Now, researchers at the University of Toronto’s Rossy Centre, an institution dedicated solely to PSP research at the , have found new evidence supporting the old idea that the key may be in the “oligomers” or “high-molecular weight tau” or “HMW tau.” These are stacks of tau protein molecules small enough to remain dissolved in the brain’s fluids, as opposed to single molecules or the large, insoluble neurofibrillary tangles visible through a conventional microscope.
The top-line result was that the patients with more rapidly-progressive PSP and brain regions with the worst damage had higher levels of HMW tau. In a tour-de-force of lab experiments, the Toronto researchers also showed that:
HMW tau was more resistant to the brain’s mechanism for breaking down such protein clusters.
The study’s 25 PSP patients could be divided into high-, medium- and low-seeders based on the speed with which their tau converted healthy tau to their own misfolded form.
Tau with phosphate groups attached to amino acids 202 and 205 were least likely to form the HMW tau clusters.
The pattern of production of proteins (i.e., the “proteomics”) in the brain areas rich in HMW tau showed disruption of the brain’s adaptive immune system and two other cellular systems previously known to be related to neurodegeneration.
The importance of all this is that we now have a more specific idea of the structure of the most toxic form of tau aggregates and that boosting the brain’s adaptive immune system with medication could discourage the seeding of misfolded tau into healthy cells.
The study’s first author, Dr. Ivan Martinez-Valbuena, published an editorial in the journal Brain Pathology explaining all this in language that non-specialist scientists can understand.
The research paper itself is posted by the authors in bioRxiv (“bio-archive”) an on-line, open-access website for articles awaiting word from the peer-review process at a conventional journal. Its senior author is Dr. Gabor Kovacs, one of the world’s leading neuropathologists in the field of neurodegenerative diseases.
A caregiver has asked me, as CurePSP’s Chief Clinical Officer, to list the most important clinical research advances in PSP of 2023. Happy to oblige. Here are my top five in no particular order.
The FDA approved a combination of two drugs called taurursodiol and sodium phenylbutyrate with the brand name “Relyvrio” for use in amyotrophic lateral sclerosis (ALS; Lou Gehrig disease). A trial in PSP has already started to recruit patients. The drugs address an issue in the mitochondria shared by the two diseases in different sets of neurons.
Tau PET ligand APN-1607 received go-ahead from the FDA to proceed to a pivotal Phase 3 trial. Such a trial began recruitment in December in the US and will involve multiple other countries as well. The compound would allow a diagnosis of PSP in early or equivocal cases by being taken up by the abnormal tau protein in the brain and imaged.
A drug called TPN-101 was found to be safe and well-tolerated in a Phase 1 trial of 30 patients with PSP. The drug counters inflammation in the brain by reducing the transcription of ancient viral DNA in our genome. Next is a small trial for efficacy.
A simple, remote, gait-monitoring system with only three sensors proved able to distinguish the gaits of PSP and PD. Further testing for its ability to document progression or improvement will follow.
PET imaging of frontal lobe synapses showed good correlation with the PSP Rating Scale and with the results of cognitive testing. This is different from typical PET in neurodegenerative disease, which images glucose utilization or protein aggregates. The work suggests that synaptic imaging could be a good diagnostic marker in the earliest, pre-symptomatic stages of PSP.
But the most important piece of news is that several drug companies are planning to start clinical treatment trials in the next year or two. I’ll report on all that as it happens.